

铱酸盐开放骨架衍生的高活性长寿命析氧电催化剂
描述
01
导读
酸性析氧反应是几种重要的电能-化学能转换的基础,这种能量密集型过程在工业上依赖于铱基电催化剂。以铱酸盐为代表的的电催化剂性能强烈依赖于初始铱酸盐的晶体结构和缺陷。最近报道的具有无定形活性相的铱酸盐衍生的电催化剂通常表现出比结晶IrO2纳米催化剂高10倍以上的活性,但结构稳定性更差(或铱浸出更严重)。并且,它们中的大多数在电催化条件下仅保持不到50小时的高催化活性。因此,希望开发新的铱酸盐(预)电催化剂,其可以避免传统催化剂中的从结晶到无定形的相变,从而实现催化活性和寿命的同时提高。
02
成果简介
鉴于此,吉林大学邹晓新教授团队报道了具有开放框架结构的亚稳态铱酸锶的相选择性合成,该开放框架铱酸盐衍生的纳米催化剂在酸中给出了与最活跃的铱基析氧电催化剂相当的催化活性,并且保持其催化活性超过1000小时。文章以题为“Highly Active, Long-Lived Oxygen Evolution Electrocatalyst Derived from Open-Framework Iridates”发表在Advanced Materials上。
03 关键创新
(1)报道的开放框架铱酸盐衍生的纳米催化剂在酸中给出了与最活跃的铱基析氧电催化剂相当的催化活性,并且保持其催化活性超过1000小时;
(2)亚稳态铱酸锶的转化包括两个主要步骤:酸中的Sr2+/H+离子交换和电催化条件下的原位结构重排;
(3)开放框架的铱酸盐具有在酸中进行快速质子交换而没有框架无定形化的能力。在酸性析氧过程中,所得质子化的铱酸盐进一步重构为超小的、表面羟基化的、(200)晶面取向的金红石纳米催化剂,而不是普通的无定形IrOxHy相。这种微结构特征有利于电催化循环中羟基的氧化和O-O键的形成。
04
核心内容解读
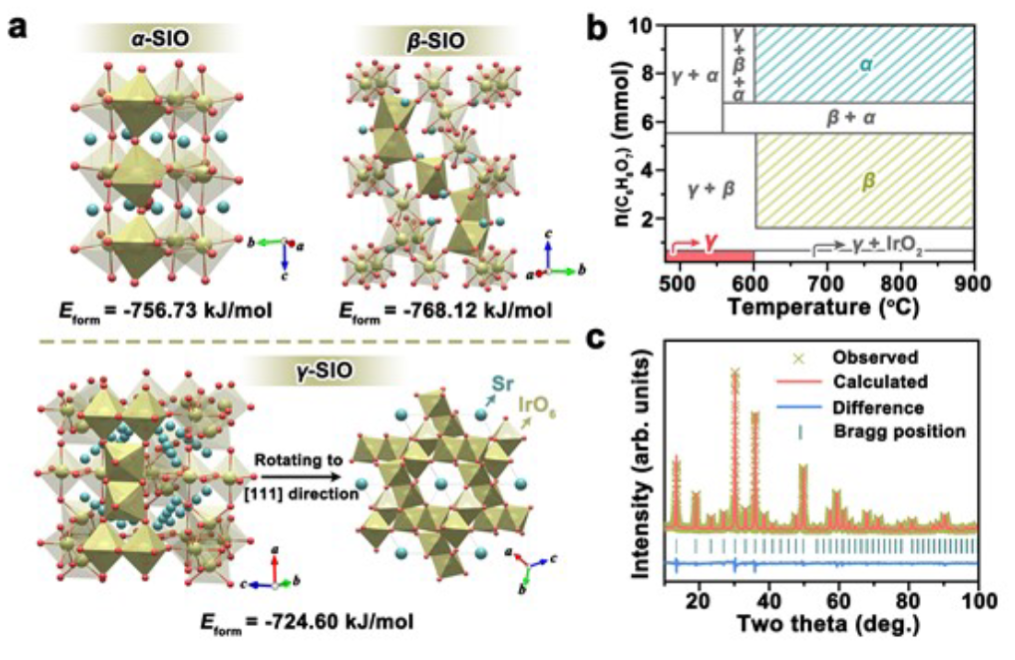
图1. (a)α-SIO, β-SIO and γ-SIO的晶体结构。(b)各种铱酸锶相的选择性合成的示意图。(c)具有γ-SIO细化图的XRD图。
图1a显示了三种铱酸锶相的晶体结构。α-SIO和β-SIO都是致密结构,γ-SIO采用三维体心开放框架结构。α-SIO仅包含角共享IrO6八面体,而β-SIO由角共享IrO6八面体和面共享IrO6八面体二聚体组成。γ-SIO由共享边缘的IrO6八面体二聚体组成。
如图1b所示,与α-SIO和β-SIO的合成相比,用少量柠檬酸和低煅烧温度实现了相纯γ-SIO的合成。粉末x射线衍射(XRD)图(图1c)证明了具有开放框架结构的高纯度铱酸锶的成功合成。
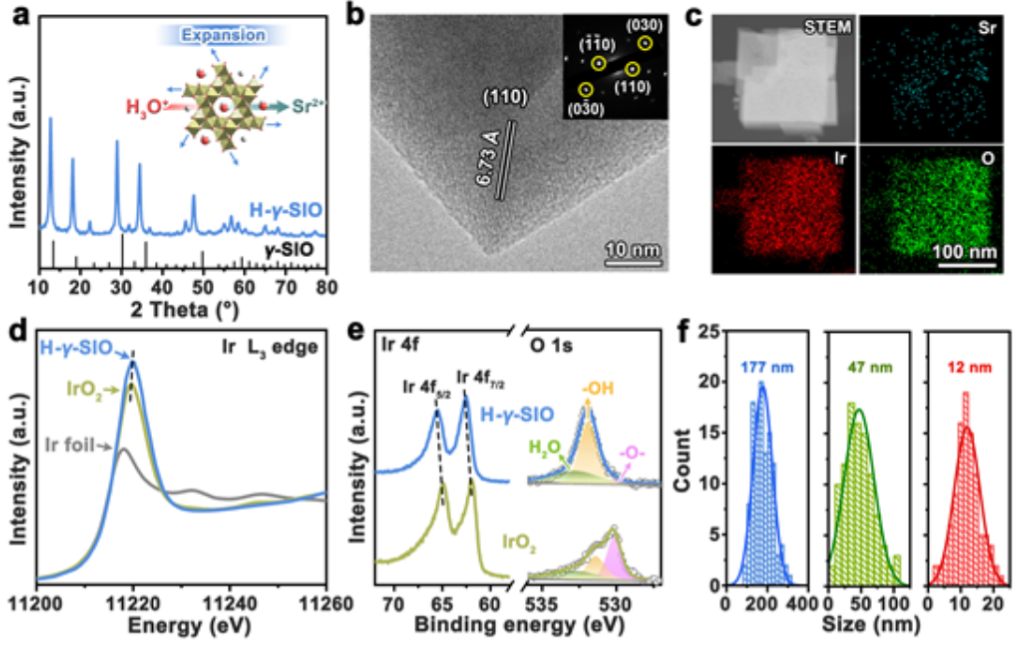
图2. (a)H-γ- SIO的XRD图。(b)H-γ- SIO的高分辨率透射电子显微镜图像。(c)H-γ-SIO的相应元素映射图像。(d)Ir箔、IrO2和H-γ-SIO的XANES光谱。(e) IrO2和H-γ-SIO的Ir 4f和O 1s XPS光谱。(f)分别为H-γ-SIO-3、H-γ-SIO-2和H-γ-SIO-1的尺寸分布。
酸腐蚀对酸性OER电催化剂的结构有很大影响。在电催化研究之前,我们首先测试了γ-SIO在酸性溶液中的结构稳定性。在酸处理后,发现开放骨架中的Sr2+离子与H+离子完全交换,产生质子化形式(记为H-γ-SIO)。
H-γ-SIO的XRD图(图2a)与γ-SIO的粉末衍射图非常吻合,表明H-γ-SIO保持了γ-SIO的开放骨架结构,但呈现出轻微的晶格膨胀。HRTEM图像 (图2b)进一步证实了晶格膨胀。H-γ-SIO的元素映射图像(图2c)显示了非常弱的Sr信号。经过酸处理后,γ-SIO中的Sr2+离子几乎完全被H+离子交换。比较了H-γ-SIO、IrO2和Ir箔的Ir L3-edge X射线吸收近边结构(XANES)光谱(图2d)。
与IrO2相比,H-γSIO中Ir原子周围的电子密度稍低。此外,相对于IrO2,H-γ-SIO的Ir 4f XPS光谱(图2e)显示出向更高结合能的小位移,并证明Ir在H-γ-SIO中的氧化态高于IrO2。与IrO2相比,H-γ-SIO具有高度羟基化的表面,这是Sr2+/H+离子交换的结果。
图2f给出了三个样品的尺寸分布和平均直径(数均)分别为177、47和12 nm。相应地,这三种样品按粒径降序分别标记为H-γSIO-3、H-γSIO-2和H-γSIO-1。
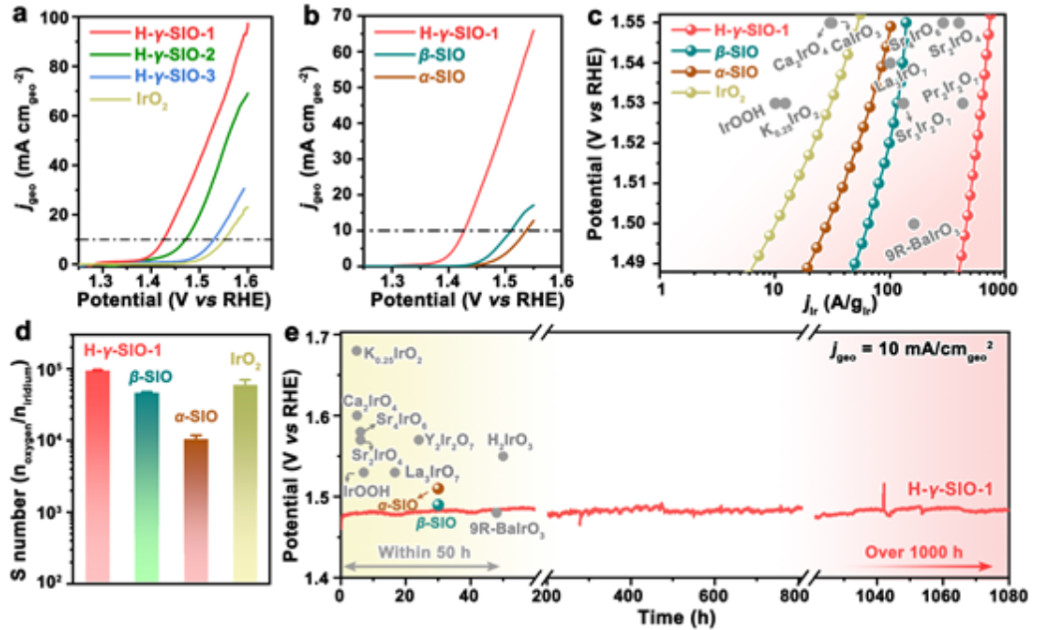
图3. (a) H-γ-SIO-1、H-γ-SIO-2、H-γ-SIO-3和IrO2作为电催化剂的OER在0.1 M HClO4中的极化曲线。(b)OER与H-γ-SIO-1、β-SIO和α-SIO在0.1 M HClO4中的极化曲线。(c)H-γ-SIO-1的Ir质量活性与一些先前报道的在酸中由铱酸盐衍生的电催化剂的比较。(d)H-γ-SIO-1、β-SIO、α-SIO和IrO2的计算S数。(e)在没有红外补偿的情况下,在10 mA cmgeo-2电流密度和H-γ-SIO-1存在时,OER的计时电位曲线。
对三种不同粒径的H-γ-SIO样品的电催化性能进行评估,并以IrO2纳米催化剂作为参比材料进行了研究。对比OER在酸中的极化曲线(图3a)。在10 mA cmgeo-2电流密度下,H-γ-SIO-1、H-γ-SIO-2、 H-γ-SIO-3和IrO2的过电位分别为200、248、270和320 mV。
如图3b所示, H-γ-SIO-1的电催化活性(通过几何电流密度评估)优于所有先前报道的铱酸盐衍生的电催化剂。进一步将测量的电流相对于铱的质量进行归一化。H-γ-SIO-1在1.5 V下给出466 A·g-1的电流密度,比IrO2(10.3 A·g-1)大45倍。H-γ-SIO-1的铱质量活性也高于那些代表性的铱酸盐基(预)电催化剂。以上结果表明,H-γ-SIO-1是一种高效的铱基氧化物电催化剂。
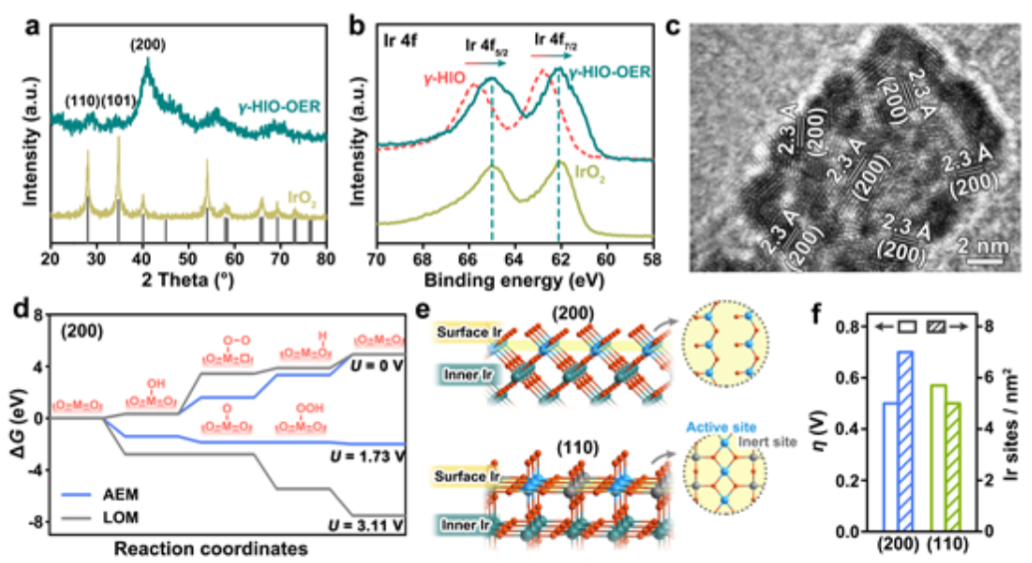
图4. (a)在酸性溶液中以10 mA/cmgeo2进行10小时计时电位测试后,H-γ-SIO-1和IrO2的粉末XRD图。(b)OER试验前后,H-γ-SIO的Ir 4f XPS光谱。(c)试验后H-γ-SIO-1的HRTEM图像。(d)通过AEM反应途径和LOM反应途径,IrO2的(200)表面在不同外加电势下的四级自由能图。(f)IrO2的(200)和(110)表面的理论超电势和I活性位数目的比较。
为了研究电催化过程中可能的结构重建,对OER后的H-γ-SIO-1进行了表征。与IrO2不同,H-γ-SIO-1的XRD图在OER后发生了显著变化催化后的H-γ-SIO-1(图4a)的衍射图与IrO2的衍射图在峰位置上相同,并且最强的衍射峰是IrO2的(200)面,而不是常见的(110)和(101)面。H-γ-SIO-1的Ir 4f XPS光谱(图4b)在OER后显示出明显的向较低结合能的移动,表明Ir的氧化态降低。催化后的H-γ-SIO-1的HRTEM图像(图4c)显示样品由晶格间距约为2.30的小纳米颗粒(1~2 nm)组成,对应于IrO2的(200)晶面。H-γ-SIO-1在催化过程中经历了重组,形成(200)取向的IrO2纳米粒子。
基于OER后的上述表征,H-γSIO-1的优异催化稳定性可归因于两个方面。一方面,结晶金红石IrO2纳米颗粒是最终的活性相,其具有高的固有稳定性和在OER过程中超低的铱浸出。另一方面,制约析氧电催化剂在酸中长期稳定性的一个重要障碍是催化剂与集流体之间的界面接触不良,导致催化剂/电极界面快速氧化钝化,容易失活。另外,部分原因可能是其超小纳米颗粒形态,改善了催化剂与集流体的界面接触。
红外基电催化剂上的OER包括两种催化机理,吸附质析出机理(AEM)和晶格氧机制(LOM)。IrO2(200)表面上AEM和LOM的反应路径的计算表明,类似于通常研究的IrO2纳米催化剂,H-γ-SIO-1的OER优选遵循AEM途径,而不是LOM途径,因为其活性相是(200)晶面取向的IrO2。
根据AEM,我们通过密度泛函理论(DFT)计算研究了IrO2的(200)面上表面Ir位的催化活性。在四步反应机理中(图4d), O–O键形成步骤(O* + H2O → OOH* + H+ + e-)是电势决定步骤,并且超电势被计算为0.5 V。金红石IrO2的(200)和(110)表面上的IR活性位点的数量(图4e)的计算结果表明,(100)表面(即(200)表面的等效面)比IrO2的热力学最稳定的(110)表面具有更高的活性。
这些结果表明,IrO2的(200)表面具有更多数量的Ir活性位点,并且每个Ir活性位点的本征活性更高(图4f)。这种不寻常的晶面取向和极小的纳米晶尺寸使得H-γ-SIO-1在电化学测试中具有优异的OER活性。
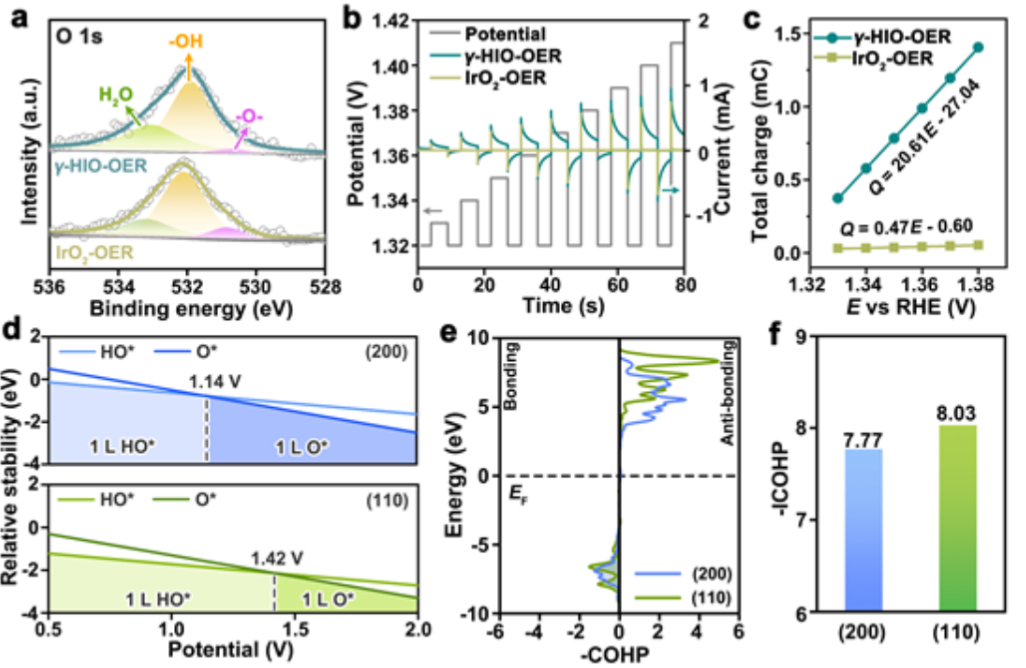
图5. (a) 实验后,H-γ-SIO-1和IrO2的O 1s XPS谱。(b)测量催化后的H-γ-SIO-1和IrO2对脉冲伏安法的电流响应。(c)储存的电荷与催化后的H-γ-SIO-1和IrO2的电势之间的关系。(d) IrO2的(200)和(110)表面的表面相图。(e)O-H键的COHP曲线和(f)IrO2的(200)和(110)表面的积分COHP值。
基于上述催化机理,O*是决定OER活性的关键中间体。对于催化后的H-γ-SIO-1和IrO2样品,它们的表面更倾向于OH-涂层。O 1s XPS分析 (图5a)反映催化剂表面储存的氧化电荷的脉冲伏安法测量可用于测量羟基的去质子化能力。在不同的电压脉冲下,催化的H-γ-SIO-1和IrO2显示出交替的阴极和阳极电流脉冲(图5b)。
通过对电压脉冲的阳极电流响应进行积分,可确定储存的氧化电荷(图5c)。催化的H-γ-SIO-1的储存电荷是IrO2的44倍。这些发现表明,与IrO2相比,催化的H-γ-SIO-1促进了O*中间体的产生,这是需要形成O-O键的限速步骤所必需的。
为了进一步理解去质子化能力,作者生成了IrO2的(200)和(110)表面的表面相图(图5d)。在低电位下,羟基封端的表面对IrO2的(200)和(110)表面都是最稳定的。然而,去质子化过程是由于在较高电位下,IrO2的(200)和(110)表面上的Ir位置优先被O*覆盖。(200)面更容易产生O*,因为它的去质子化势(1.14 V)低于(110)面(1.42伏)。
为了比较HO覆盖的(200)和(110)表面的去质子势,计算了O-H键的晶体轨道哈密顿布居(COHP)。结果(图5e)显示,O-H电相互作用由O 2p和H 1s态之间的轨道杂化主导。(200)表面的O-H杂化的积分COHP值(OH键强度的定量描述)为7.77,低于(110)表面的积分值(8.03)(图5f),进一步表明,IrO2的(200)表面的O-H相互作用更弱,更容易去质子化。
05
成果启示
开放骨架铱酸盐在酸性电解质中电催化OER 期间重建为超小的、表面羟基化的、(200)晶面取向的金红石纳米催化剂。不寻常的(200)面上高活性Ir位点的大密度,加上超小的纳米晶体尺寸,促进了对OER的优异催化活性。该发现为传统的铱酸盐电催化剂在酸性OER中面临的从晶态到非晶态的相变问题提供了解决方案,并且可以鼓励对高活性、长寿命的铱酸盐基电催化剂的进一步研究。
审核编辑:刘清
-
碱性醇类燃料电池新型催化剂的研究2011-03-11 0
-
燃料电池氧电极催化剂的研究2011-03-11 0
-
长寿命NTC热敏电阻2013-07-27 0
-
怎么对烙铁头进行的正确维护及延长寿命2010-02-27 1588
-
高效电催化二氧化碳还原反应催化剂成功研制2020-03-30 4507
-
电催化与电催化电极的原理及研究2021-02-10 2378
-
调控锂盐消除催化剂表面凝胶化构筑高比能锂硫电池2022-07-13 1265
-
Mo配位FeCoNiMo碳负载高熵电催化析氧催化剂图文解析2022-09-20 1644
-
蜂窝状多孔结晶异质电催化剂实现高效的CO2吸附/活化2022-09-30 1986
-
应变效应对催化剂活性的影响2022-10-26 1620
-
碳纳米管桥接策略用于双功能氧电催化剂2022-11-11 1028
-
“纳米岛”型催化剂突破传统催化剂活性和稳定性的矛盾2022-11-18 543
-
如何提高HEAs催化剂的催化活性和优选设计研究2022-12-14 742
-
EnSM:锂硫电池单原子催化剂的基础、应用和机遇2022-12-22 1046
-
2D催化剂层电催化活性的等离子体成像2023-01-08 735
全部0条评论

快来发表一下你的评论吧 !
