

固定电势在电化学反应中的应用
描述
应用背景
1970年,Bockris在《Mordern Electrochemistry》一书中把电化学定义为:研究带电界面的现象的科学,及研究电子导体和离子导体界面现象的科学。利用第一性原理计算探究界面反应机理,是目前广泛应用的研究手段之一。
在电化学界面反应过程中,由于电化学反应界面通常与恒定电极电势的外电极相连,为确保电子的化学势与外电极的电势达到平衡,即电子的巨正则系综(grand canonical ensemble),实际体系中会存在电子的流入与流出过程。传统的第一性原理计算通常是在正则系综下,即在电荷守恒的条件下展开的,因此它并不能很好的描述电化学界面反应,下文中我们将在电荷守恒的条件下展开的计算模型称为恒电荷模型(constant charge model,CCM)。
因恒电荷模型并不适于处理电化学界面问题,我们可采用在电子巨正则系综下展开第一性原理计算,这种计算方法又被称为固定电势方法(fixed potential method/constant potential method)。在下文中我们将利用固定电势计算的模型称为恒电势模型(constant potential model,CPM)。
后文提供了使用DS-PAW计算电催化氮还原反应(Electrocatalytic nitrogen reduction reaction, eNRR)过程中反应能计算的详细教程。
案例概览
本文展示了如何使用DS-PAW模拟一个电催化还原氮气反应。该反应以碳基负载过渡金属Ru单原子为催化剂,使用DS-PAW对电催化 N2分子的吸附及还原过程进行模拟。在计算过程中使用了CCM_vacuum,CCM_water,CPM_water三种不同的模型,本文详细介绍了在不同模型下如何设置参数及计算反应能。
N2分子的吸附及还原过程进行模拟。在计算过程中使用了CCM_vacuum,CCM_water,CPM_water三种不同的模型,本文详细介绍了在不同模型下如何设置参数及计算反应能。
更多关于DS-PAW的使用细节,请移步DS-PAW手册查看。
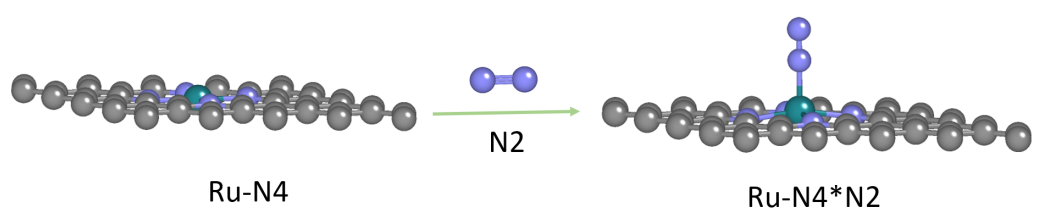
图1. 吸附过程示意图
计算流程
DS-PAW模拟的反应为 N2 分子在碳基负载Ru单原子上的吸附过程,反应的表达式可简写作:(Ru − N4 ) + N2 = (Ru − N4 − N2 )
第一步:搭建模型
模型包括:(a) 碳基负载Ru原子模型(Ru − N4) ,(b) N2 单分子模型,(c) 吸附了 N2 分子的碳基负载Ru原子模型(Ru − N4 − N2)
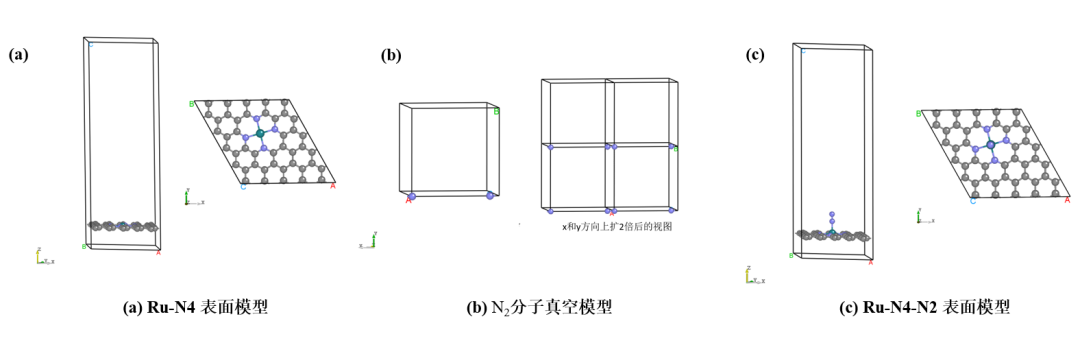
图2. 计算模型图
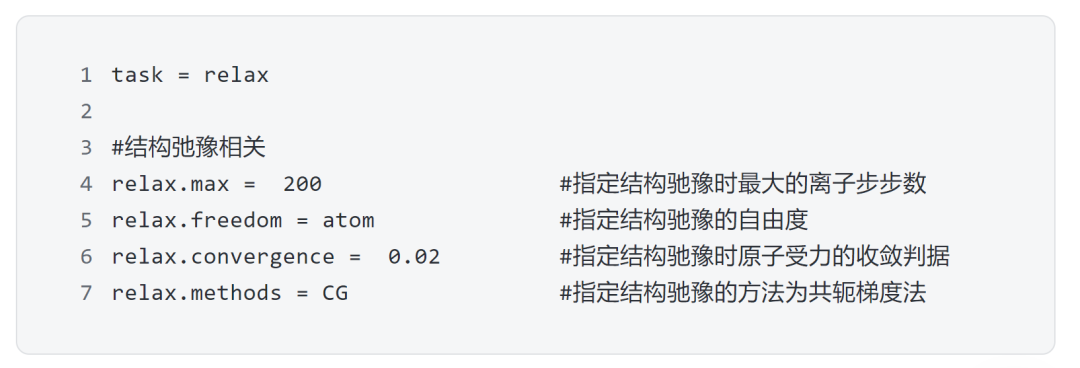
第二步:结构弛豫
对搭建的结构进行结构弛豫,获得稳定结构,在DS-PAW中进行结构弛豫需要的核心参数:
第三步:能量计算
在不同模型条件下进行能量计算,获得稳定构型对应能量,下面按不同模型分别进行介绍:
CCM_vacuum
常规的真空层下的scf计算,即可获得CCM_vacuum模型下的能量,下面列出了在DS-PAW中进行单点能计算需要设置的核心参数:

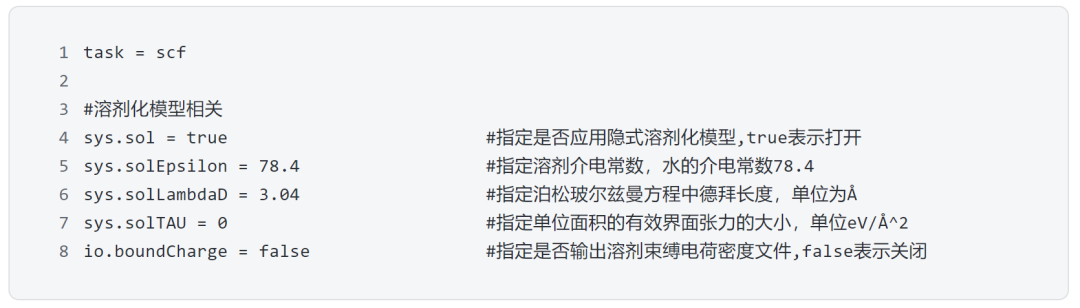
CCM_water
在CCM模型下,也可以利用隐式溶剂化模型来考虑溶剂效应,这里我们以水溶液为例,列出利用DS-PAW在scf计算中引入溶剂化模型所需要设置的核心参数:
CPM_water
在DS-PAW中用固定电势方法计算即可获得CPM模型下的能量。在新发布的2023A版本中进行固定电势计算必须引入溶剂化模型,这里列出了利用DS-PAW在隐式水溶液环境下进行固定电势计算的核心参数:

第四步:反应能计算
本文选取了3个不同的计算模型,首先介绍各模型下的吸附反应式:
CCM_vacuum:
该模型下,吸附反应式可写作:
(Ru-N4)+N2 (ideal gas)=(Ru-N4-N2)
我们定义ΔE为反应能,反应能的计算表达式为:
ΔE=E0(Ru-N4-N2)-E0(Ru-N4)-E0(N2)
其中,E0对应真空模型下体系的总能((sigma→0),该数值可从自洽计算所得的scf.h5(或system.json)文件中获取,查找关键字 “TotalEnergy0” 即可。
CCM_water:
该模型下,吸附反应式可写作:
(Ru-N4)(in water)+N2 (ideal gas)=(Ru-N4-N2) (in water)
ΔE=E0(Ru-N4-N2)-E0(Ru-N4)-E0(N2)
其中,E0对应水溶液浸润的模型下体系的总能((sigma→0),该数值可从自洽计算所得的scf.h5(或system.json)文件中获取,查找关键字 “TotalEnergy0” 即可。
CPM_water 该模型下模拟的反应过程为气相中的N2在由水溶液浸润并与0V vs. SHE(标准氢电极)电极接触的催化剂表面的吸附过程,此时吸附反应式有两种写法,为便于描述,我们定义了以下物理量符号:
ne0 : 中性体系下的核电荷数
ne : 当体系电压为设定值(sys.fixedPPotential所设数值,此例对应 0 V)时体系的总电子数
dne : 当体系电压为设定值时,体系的带电量:dne = ne − ne0
μe : 体系电子化学势,电势零点为溶液深处(即DFT计算得到的电荷密度最低点的电势值)
Δe : 吸附态体系价电子数(eAB)与吸附基底和吸附分子总价电子数(eA+eB)的差值
Ω0 : grand total energy(sigma→0): 电子巨正则系综下的体系总能,其表达式为:Ω0 = E0 − dne ∗ μe
此时,CPM_water模型下吸附反应式可参考如下写法:
方法一、在反应式中考虑 Δe ,吸附反应式可写作:
Ru-N4 (0V vs. SHE) + N2(ideal gas) = Ru-N4-N2 (0V vs. SHE) - Δe
ΔE = E0(Ru-N4-N2) − Δe * μe − E0(Ru-N4) − E0(N2)
其中,E0的数值可从自洽计算所得的 scf.h5(或system.json)文件中获取,查找关键字 “TotalEnergy0” 即可。
ne 和 μe 的数值可从自洽计算所得的 DS-PAW.log (或 scf.h5 或 system.json)文件中获取,最后一个LOOP下查找关键字 “Electron” 和 “Chemical Potential(electron) ” 即可。
方法二、考虑电子巨正则系综下的体系总能 Ω0
由于固定电势计算是模拟的电子的巨正则系综,此时反应能计算式中的体系总能 E0 应当用 Ω0 来代 替。吸附反应式可写作:
(Ru-N4) (0V vs. SHE) + N2(ideal gas) = (Ru-N4-N2) (0V vs. SHE)
ΔE = Ω0 (Ru-N4-N2) − Ω0 (Ru-N4) − Ω0 (N2)
其中,Ω0 的数值可从自洽计算所得的DS-PAW.log (或 scf.h5 或 system.json)文件中获取,最后一个LOOP下查找关键字 “Grand Total Energy” 即可。
由于(Ru-N4)与(Ru-N4-N2)的电势为0V vs. SHE,故对(Ru-N4)与(Ru-N4-N2)进行0V下的固定电势计算,从DS-PAW的相应输出文件提取数据,得到如下表格,能量单位为 eV:
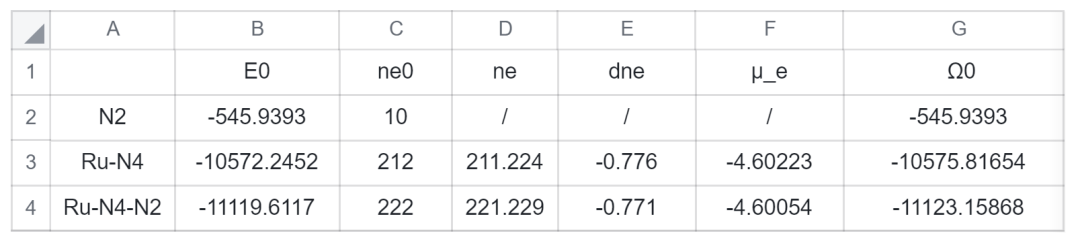
表 1. CPM_water模型下固定电势计算数据
接下来将表1的数据代入对应的表达式中进行计算:
方法一、在反应式中考虑 Δe ,反应能计算过程如下:
Ru-N4 (0V vs. SHE) + N2(ideal gas) = Ru-N4-N2 (0V vs. SHE) - Δe
ΔE = E0(Ru-N4-N2) − Δe * μe − E0(Ru-N4) − E0(N2)
= − 11119.6117 − (221.229 − 211.224 − 10) × ( − 4.600) − ( − 10572.2452) − ( − 545.9393)
= − 1.4042 eV
方法二、考虑电子巨正则系综下的体系总能 Ω0 ,反应能计算过程如下:
(Ru-N4) (0V vs. SHE) + N2(ideal gas) = (Ru-N4-N2) (0V vs. SHE)
ΔE = Ω0 (Ru-N4-N2) − Ω0 (Ru-N4) − Ω0 (N2)
= − 11123.1586 − ( − 10575.8165) − ( − 545.9393)
= − 1.4027 eV
通过两种方法计算所得的吸附能一致。可见⽤DS-PAW中定义的Ω0即可很方便的计算固定电势下的反应能。
结果展示
将表1的数据分别代入CCM_vacuum、CCM_water、CPM_water模型的吸附反应式,计算三个模型下,eNRR前三步反应的反应能,结果如下表2所示:
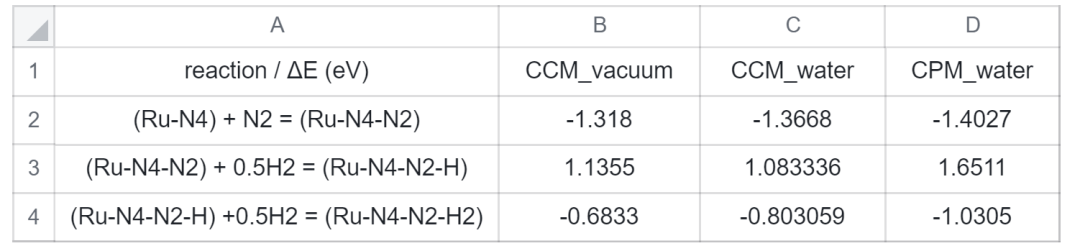
表 2. 反应能数据表
最后将上述结果绘制成反应坐标曲线,效果如下图3所示:
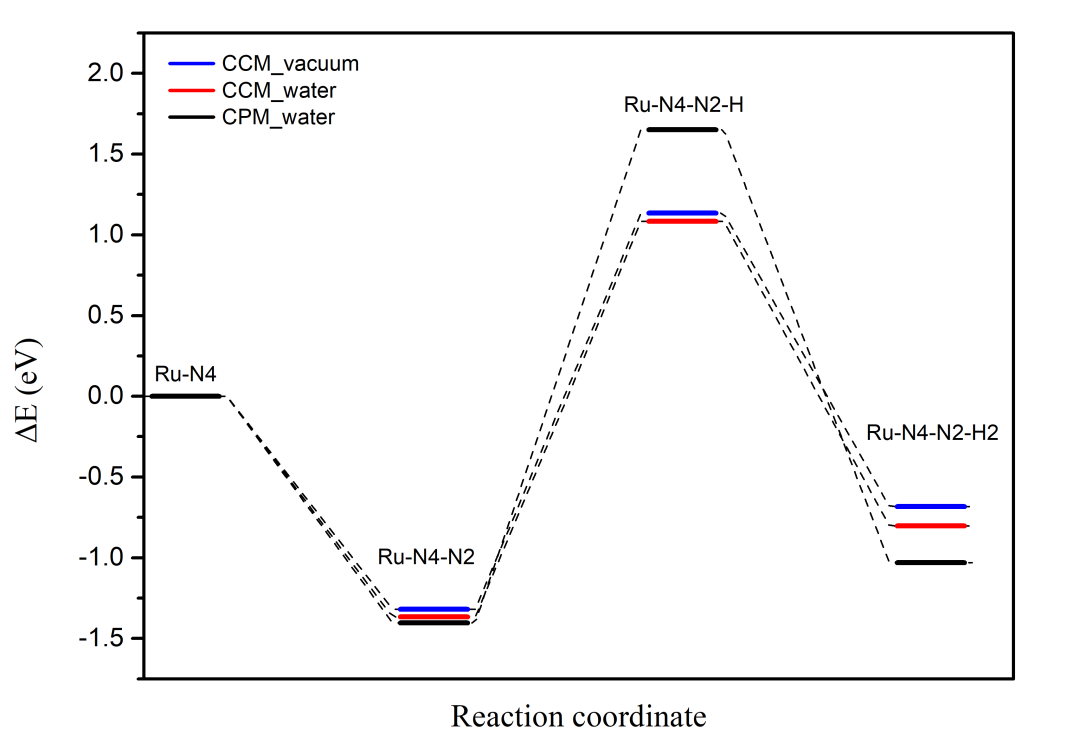
图 3. 反应坐标-反应能曲线
审核编辑:刘清
- 相关推荐
- CPM
-
电化学传感器ppt2008-07-02 0
-
采用循环伏安法研究镍在碱性溶液中的电化学活性2017-09-15 0
-
电化学原理介绍和分析方法2017-10-16 0
-
应用电化学传感器的设计2018-11-15 0
-
基于电化学传感器的Arduino兼容电化学气体检测电路CN03572020-03-12 0
-
电化学传感器的发展怎么样?2020-03-25 0
-
电化学工作站有什么功能?2020-03-30 0
-
电池的电化学阻抗谱原理是什么2021-03-11 0
-
熔盐电化学反应炉温度控制系统研究2009-02-27 661
-
燃料电池内的电化学反应-触媒与反应2009-11-02 578
-
氧化还原反应与电化学2009-11-03 489
-
电化学的应用领域2009-11-05 8035
-
什么是电化学储能?电化学储能技术主要包括哪些?2024-04-26 80
-
电化学储能的特点包括哪些?电化学储能的效率?2024-04-26 90
-
压缩空气储能属于电化学储能技术吗2024-04-26 79
全部0条评论

快来发表一下你的评论吧 !
