

莫一非团队揭示固态电池中锂金属结晶机理
描述
【研究背景】
结晶是材料科学、物理和化学中的一个重要现象。当前人们主要研究由温度或溶液变化引起的结晶过程,电化学沉积下的结晶过程仍然很少被研究,特别是下一代高能可充电电池中Li、Na、Mg和Zn金属负极的电化学沉积和结晶过程依然鲜为人知。
在电化学沉积过程中,电解质中的金属离子沉积并结晶为金属晶体。过高的结晶能垒会提高电化学沉积的过电位。高过的电位或极化会降低电池性能和效率,甚至导致锂枝晶的生成。通过降低过电位可以提高金属负极的电化学性能。使用固态电解质来解决目前困扰金属负极的问题是一个很有前途的方向。
固态电解质中金属负极的电化学沉积行为跟与液体电解质中完全不同。在液体电解质中,电化学沉积过程中金属颗粒的形核生长过程可以用经典的成核理论来描述。相比之下,在固态电解质上的锂连续沉积过程中,这些沉积的锂离子如何成为锂金属结晶的路径和机理仍然不清楚。这种结晶过程具有固有的势垒,并且会限制的电化学金属沉积过程的速率。进一步改进金属负极(如锂金属负极)的性能,急需从原子尺度上揭示结晶的原子路径和动力学势垒。
【工作简介】
近日,美国马里兰大学莫一非教授团队采用大规模分子动力学模拟方法,研究并揭示了固体界面处锂结晶的原子路径和能垒。研究发现,锂结晶采用由界面锂原子介导的多步路径,具有无序和随机密堆积构型作为结晶的中间步骤,这产生了结晶的能垒。这种对多步结晶路径的理解将奥斯特瓦尔德分步规则(Ostwald's step rule)的适用性扩展到界面原子态,并提出了将有利的界面原子态作为中间步骤来降低结晶势垒的合理策略。本文作者的研究结果开辟了界面工程的全新研究途径,这将促进固态电池金属电极的电化学沉积和结晶过程。该文章以“Lithium crystallization at solid interfaces”为题在线发表在国际顶级期刊Nature Communications上。现任同济大学材料科学与工程学院特聘研究员杨孟昊是本文第一作者。
【内容表述】
为了研究固态电解质界面上金属负极的电化学沉积行为。原子尺度建模方法在揭示固态电解质界面上原子转变机制方面具有独特的优势,可以表征每个原子的能量并达到飞秒级别的实时分辨率。在本项研究中,本文作者使用固态电解质界面处的锂金属负极作为系统模型,采用大规模分子动力学模拟(MD)方法,以直接揭示固态电解质界面上锂连续沉积过程中结晶的原子路径和动力学势垒。
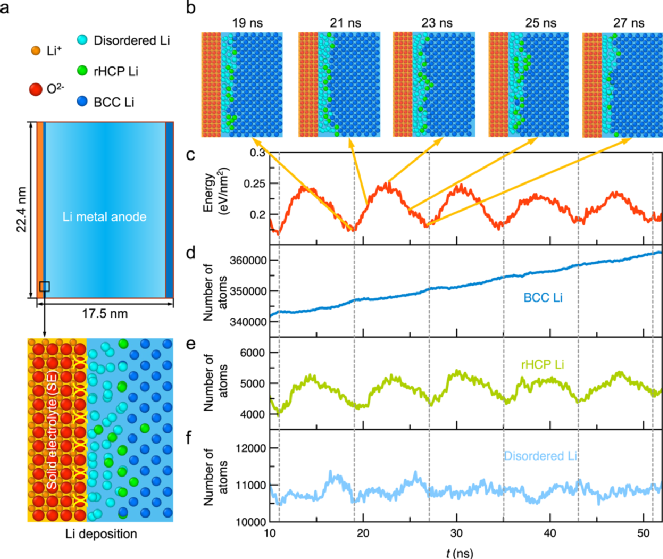
图1. 锂沉积过程中固态电解质界面锂结晶的原子模型。a)分子动力学模拟中包含锂金属层(蓝色)和固态电解质层(橙色)的原子模型;b)锂沉积过程中一个能量变化周期下的Li-SE界面的原子结构;在锂沉积过程中,c)以体相锂金属为参考时锂金属层的能量;d-f)锂金属层中不同局域构型下锂原子的数量(如:体心立方(BCC)、随机密排六方相(rHCP))。
Li-SE界面上界面原子结构在锂电池循环过程中对金属锂的结晶过程中起着关键作用。通过采用分子动力学模拟方法跟踪锂随时间的演变过程,作者进一步揭示了锂从锂离子到体心立方BCC-Li的逐步结晶过程。沉积的锂原子首先被Li-SE界面上非晶锂原子层吸附,随后在后续的锂沉积过程中通过两条途径结晶成BCC锂金属过程。
在第一种途径中,沉积的Li原子首先通过disordered-Li,随后转变为BCC锂金属;在第二种途径中,沉积的锂原子通过disordered-Li之后,大部分的disordered-Li会形成下一个rHCP-Li中间相,最后转变成为BCC-Li金属(图2b和d)。因此,锂结晶由固态电解质界面处非晶原子层介导,其中界面原子、disordered-Li和rHCP-Li充当多步骤结晶过程的中间结构,而这些界面原子结构是由于固态电解质和锂金属之间界面相互作用引起的。
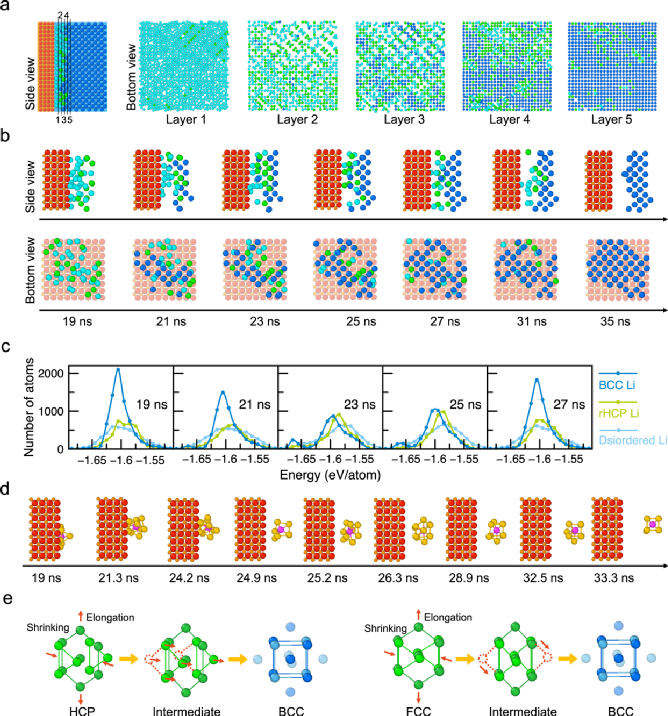
图2. 锂结晶的多步路径。a) 19 ns时Li-SE界面的原子结构,以及展示的每层原子(无序disordered、随机密排六方相rHCP、体心立方相BCC锂分别以青色、绿色、蓝色表示);b)一群锂原子的结晶过程;d) 单个锂原子(紫色)与它的近邻原子们(黄色);c)锂原子态密度(DOAS)展示了不同类型锂的原子能量分布;e)密排六方相HCP或面心立方相FCC构型向体心立方相BCC构型演变的示意图。
随机密排六方相rHCP-Li构型 (HCP 或 FCC 的混合物) 通过小的锂原子运动转变为 BCC-Li,如图2e所示。当HCP-Li转变为BCC-Li时,{0001}六角面通过向<110>方向收缩或向<001>方向伸长变为{110}面,其他原子平行于六角面沿 <110> 方向移动形成 BCC构型。
FCC-Li以类似的方式通过小的锂原子运动转变为BCC-Li(图2e)。除了具有较低的能量外,从rHCP-Li到BCC-Li的轻松转变也使rHCP-Li成为锂结晶过程中有利的中间态。这种原子路径遵循奥斯特瓦尔德分步规则,即在最终稳定状态之前形成更高能量但动力学上有利的中间态(图3)。
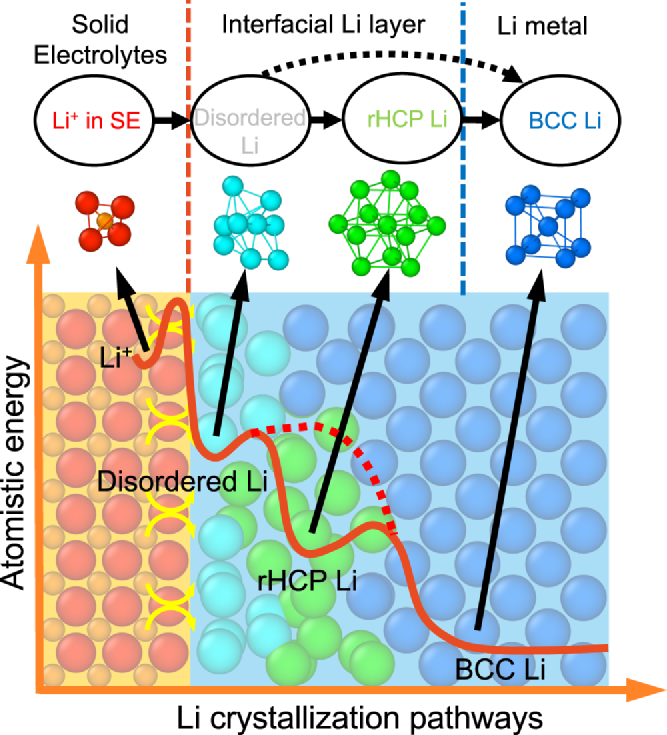
图3. 锂结晶的多步路径示意图。固态电解质中的Li+(桔色)先转变为界面层中disordered-Li(青色)以及rHCP-Li(绿色),最后转变为BCC-Li(蓝色)。
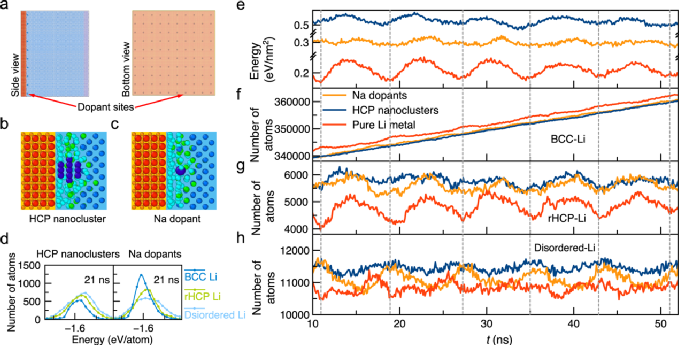
图4. Li-SE界面处的锂结晶。a)具有纳米团簇和掺杂剂的Li-SE界面模型;具有b)HCP-Li纳米团簇和c)钠掺杂剂(深蓝)的Li-SE界面的原子结构;d)不同结构类型锂的原子能;e)具有纳米团簇、掺杂剂和锂金属体相的锂能量变化;f-h)锂金属中不同近邻结构构型下锂原子数目;原始的Li-SE界面(红色)、有钠掺杂时的界面(橘色)、有HCP-Li纳米团簇时的界面(蓝色)。
【核心结论】
分子动力学模拟结果揭示了固态电解质界面处锂结晶的多步骤原子路径,这表明奥斯特瓦尔德分步规则已经扩展到单个原子态。奥斯特瓦尔德分步规则表明:在结晶过程中,在热力学稳定相之前首先形成较高能量的中间相。在锂结晶的多步原子路径中(图3),高能界面原子态(例如disordered-Li和/或rHCP-Li)首先作为中间体形成,遵循奥斯特瓦尔德分步规则,然后转变为体相晶体原子(即BCC-Li)。在这个复杂的多步结晶过程中,这些界面原子态的动力学和能量学可以通过界面原子的原子态密度(DOAS)来阐明。
作为结晶路径中间体的界面原子态是锂金属和固态电解质之间界面相互作用引起的,因此可以通过界面工程进行调整。相比之下,在液体电解质中,结晶是由形核粒子和表面原子介导的,例如表面上的吸附原子或空位,如 Terrace-Ledge-Kink模型所示。这种从界面原子态的角度对多步结晶路径的理解可以提出通过固态电解质的界面工程促进结晶的合理策略。这些调整结晶原子路径的界面工程策略为提高固态金属电池金属负极的电化学沉积性能提供了机理解释和理论指导。
审核编辑:刘清
-
锂金属电池重大突破:10分钟完成充电,可循环至少6000次北京中科同志科技股份有限公司 2024-01-10
-
锂金属电池重大突破:10分钟完成充电深圳市浮思特科技有限公司 2024-01-10
-
CR2032 pdf 3V纽扣电池中文资料2015-05-28 0
-
苹果的新专利--全固态电池2015-12-23 0
-
新技术:锂空气电池是否能成为下一代电池技术标准?2016-01-11 0
-
科普:锂空气电池是什么?2016-01-11 0
-
解密:锂空气电池2016-01-12 0
-
锂空气电池的研究进展和最新情况2016-01-13 0
-
2016年十大锂电池技术突破2016-12-30 0
-
对于锂电池的开发将面临这样的挑战2017-01-17 0
-
锂电池与铅酸电池的不同之处2018-03-31 0
-
锂空气电池未来或击败锂离子电池2018-10-09 0
-
锂二氧化锰电池有什么特点?2020-03-10 0
-
全固态锂电池中锂枝晶的生长及抑制机理的研究分析2020-04-26 7177
-
同步辐射CT技术揭示正负极串扰失效机理:不均匀的离子通量2023-05-04 1013
全部0条评论

快来发表一下你的评论吧 !
