

详细描述LAMMPS在Device Studio中的应用
描述
8.2. LAMMPS 实例
LAMMPS 即 Large-scale Atomic/Molecular Massively Parallel Simulator , 大规模原子分子并行模拟器 ,主要用于 分子动力学 相关的一些计算和模拟工作。一般来讲,分子动力学所涉及到的领域,LAMMPS代码也都涉及到了。LAMMPS由美国Sandia国家实验室开发,以GPL license发布,即开放源代码且可以免费获取使用,这意味着使用者可以根据自己的需要自行修改源代码。LAMMPS 程序在模拟固态材料(金属、半导体)、柔性物质(生物分子、聚合物)、粗粒度介观体系等方向具有广泛的应用。LAMMPS 程序内置多种原子间势(力场模型),可以实现原子、聚合物、生物分子、固态材料(金属、陶瓷、氧化物)、粗粒度体系的建模和模拟。该程序即可以模拟二维体系,也可以模拟三维体系,可以模拟多达数百万甚至数十亿粒子的分子体系,并提供支持多种势函数,具有良好的并行扩展性、模拟效率高、计算时间短等优点。
分子动力学模拟 ( Molecular Dynamics , MD )是近年来飞速发展的一种分子模拟方法,已经被广泛应用于化学化工、材料科学与工程、物理、生物医药等科学和技术领域,起到越来越重要的作用。MD模拟用来研究不能用解析方法来解决的复合体系的平衡性质和力学性质,用来搭建理论和实验的桥梁,在数学、生物、化学、物理学、材料科学和计算机科学交叉学科占据重要地位。
鸿之微科技(上海)股份有限公司在 Device Studio 2021B 中开发了适用于分子动力学计算软件 LAMMPS 的计算模块。使用Device Studio,用户可在其图形界面中方便快捷的搭建或导入计算所需的结构,并可在3D显示区域查看其结构的3D视图。搭建好结构后,用户可在LAMMPS计算模块,根据计算需要,在简洁友好的界面中设置参数 生成计算所需的输入文件 ,之后连接装有LAMMPS的 本地电脑或远程服务器 进行相关计算,在计算过程中可实时监测任务的计算状态,计算完成后可对LAMMPS的计算结果进行可视化分析。
目前用户可通过Device Studio生成LAMMPS以下计算输入文件的生成: 结构弛豫、热力学性质(热膨胀系数、体积热容、等压热容)、输运性质(均方位移、速度自相关函数、热导率)、力学性质(杨氏模量、剪切模量)、淬火、退火模拟 ;结合OVITO软件可对LAMMPS计算结果进行结构特征数据分析。
以 对金属铝沿X轴方向以一定的恒定应变速率拉伸的形变模拟 为例来详细描述 LAMMPS 在Device Studio中的应用。
8.2.1. LAMMPS计算流程
LAMMPS分子动力学计算在Device Studio中的流程如图8.2-1所示。
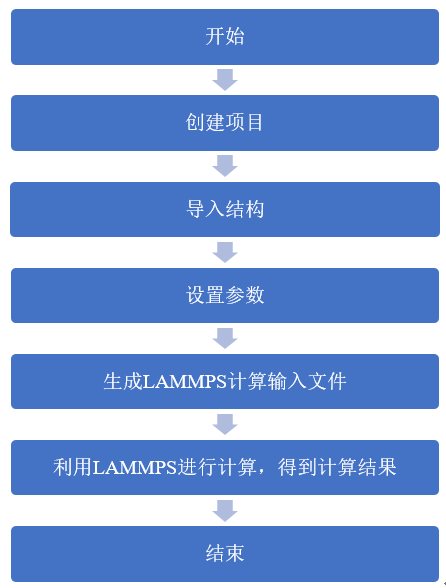
图8.2-1: LAMMPS计算流程
8.2.2. LAMMPS创建项目
双击 Device Studio 图标快捷方式启动软件,根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 LAMMPS ,或采用软件默认项目名。
8.2.3. LAMMPS导入结构
在Device Studio的图形界面中点击 File → Import → Import Local, 则弹出导入LAMMPS结构文件的界面,根据界面提示找到 Al_Rede.hzw 结构文件的位置,选中 Al_Rede.hzw 结构 文件,点击 打开 按钮则导入 Al_Rede.hzw 结构后的Device Studio界面如图8.2-2所示。
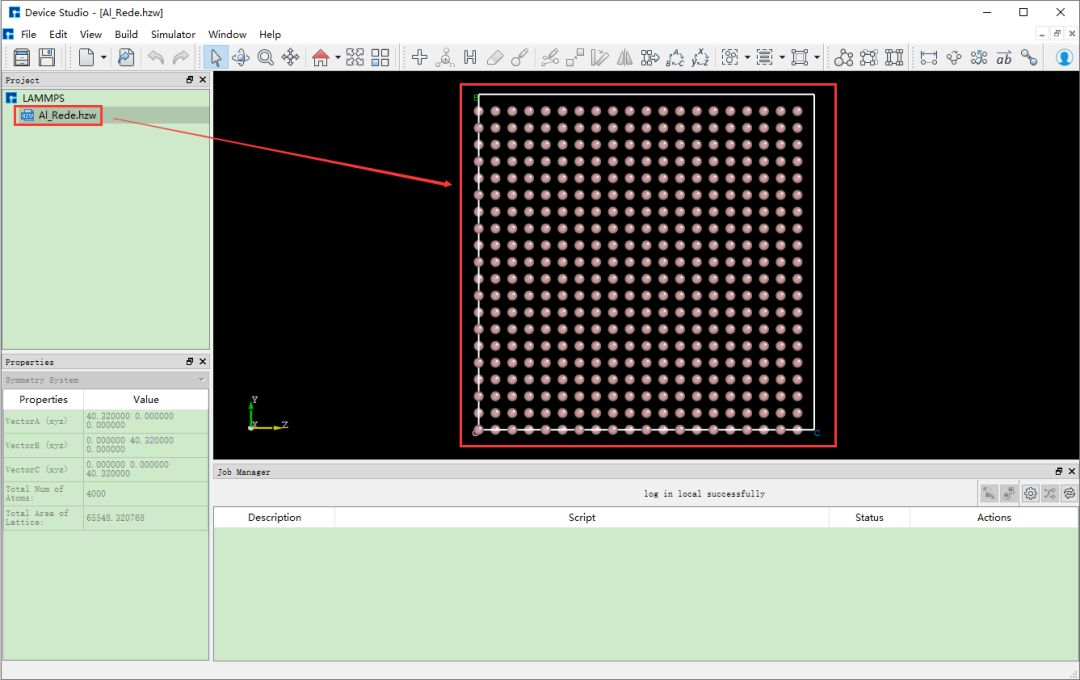
图8.2-2: 导入 Al_Rede.hzw 结构后的Device Studio图形界面
审核编辑:刘清
-
请问ad9361的寄存机详细描述在哪里?2018-08-16 0
-
哪里有ps7-gpio的设备树文档的不同属性的详细描述?2019-11-05 0
-
RTC实时时钟详细描述2022-01-13 0
-
DS1302芯片详细描述2022-01-24 0
-
详细描述RTT QSPI驱动配置过程2022-06-16 0
-
有谁知道哪里有Sequencer的详细描述以及示例?2023-01-12 0
-
多层印刷电路板及产品详细描述2009-12-10 986
-
Device Studio应用实例之LAMMPS应用实例2022-07-21 2737
-
浅谈Device Studio亮点功能2022-07-22 1281
-
Device Studio应用实例之Nanodcal应用实例2022-07-26 1126
-
Device Studio应用实例之MOMAP2022-08-03 1509
-
Device Studio应用实例之VASP(上)2022-08-04 1718
-
Device Studio应用实例之VASP(下)2022-08-04 1808
-
Device Studio应用实例之BDF(上)2022-08-12 1212
-
Device Studio应用实例之BDF(下)2022-08-14 1169
全部0条评论

快来发表一下你的评论吧 !
