

BeN4单层作为钾离子电池的优秀狄拉克阳极材料
电池技术
描述
01 引言
在全球环境的推动下,锂离子电池(LIBs)由于其容量大、能量密度高、稳定性好、使用寿命长等特点,几十年来一直在高速发展。然而,随着研究的进一步深入,LIBs的缺点已经暴露出来,例如:高成本和固有的安全问题。反过来,许多研究人员将注意力转向了其他金属离子电池(MIBs),例如钠离子电池(SIBs) 和钾离子电池(PIBs)。由于这些碱金属的性质非常相似,而且钠、钾资源更加丰富,使得SIBs和PIBs也取得了长足的进步。此外,我们知道对于PIBs来说,K+/K的标准电极电位低于Li+/Li的,因此显示出了较高的电压和能量密度。故PIBs已然成为LIBs最有希望的替代品之一。不幸的是,当经典的负极材料——石墨用于PIBs时,其存储容量仅为279mAh g-1,这远低于研究人员的预期。故迫切需要开发用于PIBs的高存储容量负极材料。二维 (2-D) 材料的特殊结构和有趣的电子特性使其具有异常丰富的特性。例如,巨大的比表面积和宽阔的暴露表面为离子扩散和存储提供了极大的便利,这表明二维材料本质上是优异的储能材料。到目前为止,许多二维材料已在理论上被证实是PIBs的良好电极候选材料,如GaS、Ti3C2、PC6、MoN2等。可惜的是,这些材料中没有一种同时具有高存储容量和低扩散势垒。因此,寻找具有许多优异性能且具有良好循环稳定性的PIBs电极材料是当前的研究热点。
02 成果简介
快速开发具有良好的稳定性、高容量、低扩散阻隔和优良循环性的阳极材料是当今进一步提高电池工业的一个重要挑战。最近,M. Bykov等人通过实验合成了一种新型的稳定的二维(2-D)材料--铍青铜(BeN4)。考虑到该二维材料的轻质和多孔结构,河北工业大学陈贵锋课题组基于第一原理计算方法,系统地研究了BeN4单层作为金属离子电池(MIBs)阳极材料的可行性:研究了Li/Na/K在BeN4单层表面的吸附行为,发现具有Dirac锥的BeN4单层只能稳定地吸附钾,并且在吸附钾之前和之后都具有良好的导电性。更值得注意的是,BeN4单层具有相当低的扩散势垒(62 meV)和开路电压(0.084-0.179 V),以及非常高的存储容量(842 mA h g-1),这些都优于大多数用于钾离子电池(PIB)的二维阳极材料。此外,BeN4单层也被证明具有很大的循环稳定性和长期的循环寿命。因此,这些结果表明BeN4单层有望成为一个相当完美的PIBs狄拉克阳极材料。
03 图文导读
图1显示了BeN4单层的3×3超胞结构的顶视图和侧视图。从侧面看,BeN4单层是平的,没有任何皱纹。从顶视图,BeN4单层是由5元和6元的环连接组成。5元环包含一个铍(Be)原子和四个氮(N)原子,而6元环包含两个Be原子和四个N原子。浅绿色的区域代表BeN4单层的原胞,它包含一个Be原子和四个N原子。从其声子谱图和分子动力学模拟图可以发现,BeN4单层具有良好的动力学稳定性和热稳定性。最后,从其能带图中可以发现,BeN4单层是半金属材料,在其费米能级附近有一个狄拉克锥。
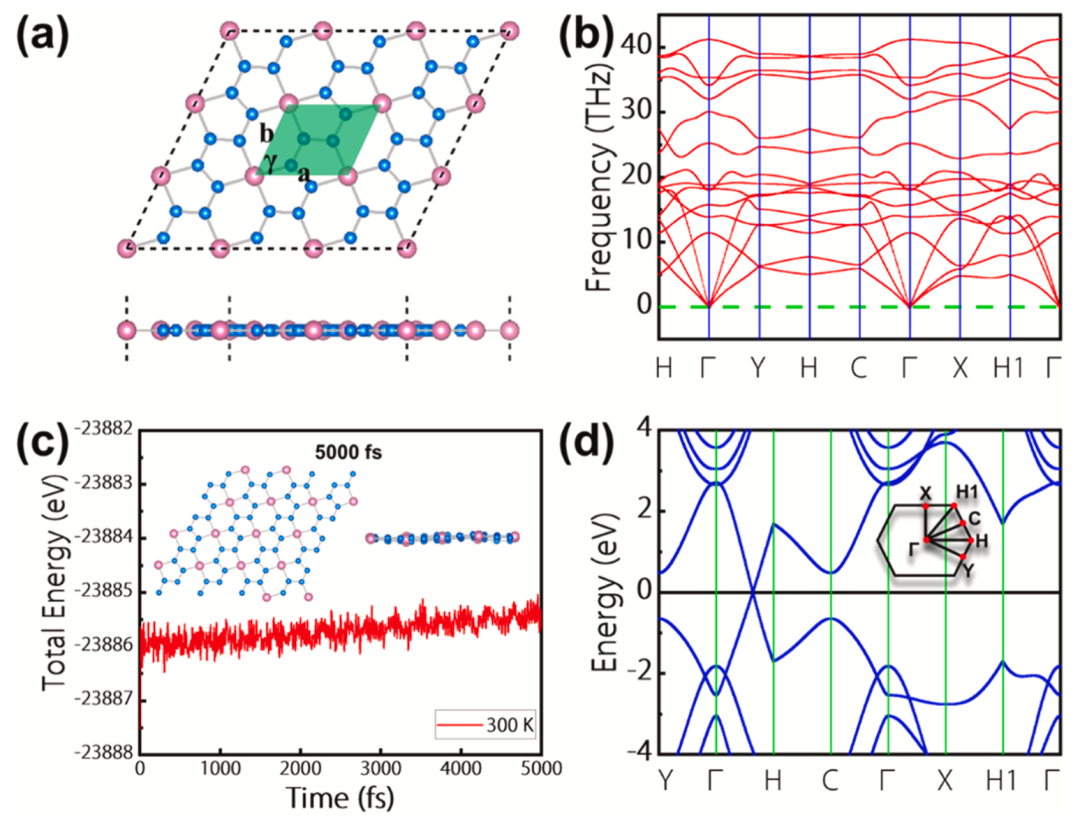
图1. (a)BeN4单层的3×3超胞的顶视图和侧视图。粉红色和蓝色的小球分别代表Be原子和N原子。在俯视图中,浅绿色区域代表BeN4单层的原胞。(b)BeN4单层的声子谱。(c)经过温度为300 K,时间为5000 fs的AIMD模拟(NVE)后的BeN4单层(4×4超胞)的能量变化和结构快照图。(d)BeN4单层的能带图。
图2显示了3×3超胞结构的BeN4单层(H1位点和H2位点)分别对碱金属(Li/Na/K)的吸附行为,发现BeN4单层只能稳定地吸附K而不能吸附Li/Na,还发现Li/Na/K在BeN4单层上同一吸附位点的吸附高度依次增加,这可能是由于Li/Na/K的半径增大。此外,Li/Na/K在H1位点的吸附高度低于H2位点的吸附高度,这一现象可以解释为对于同一类型的原子,吸附能越强,其吸附高度越低。
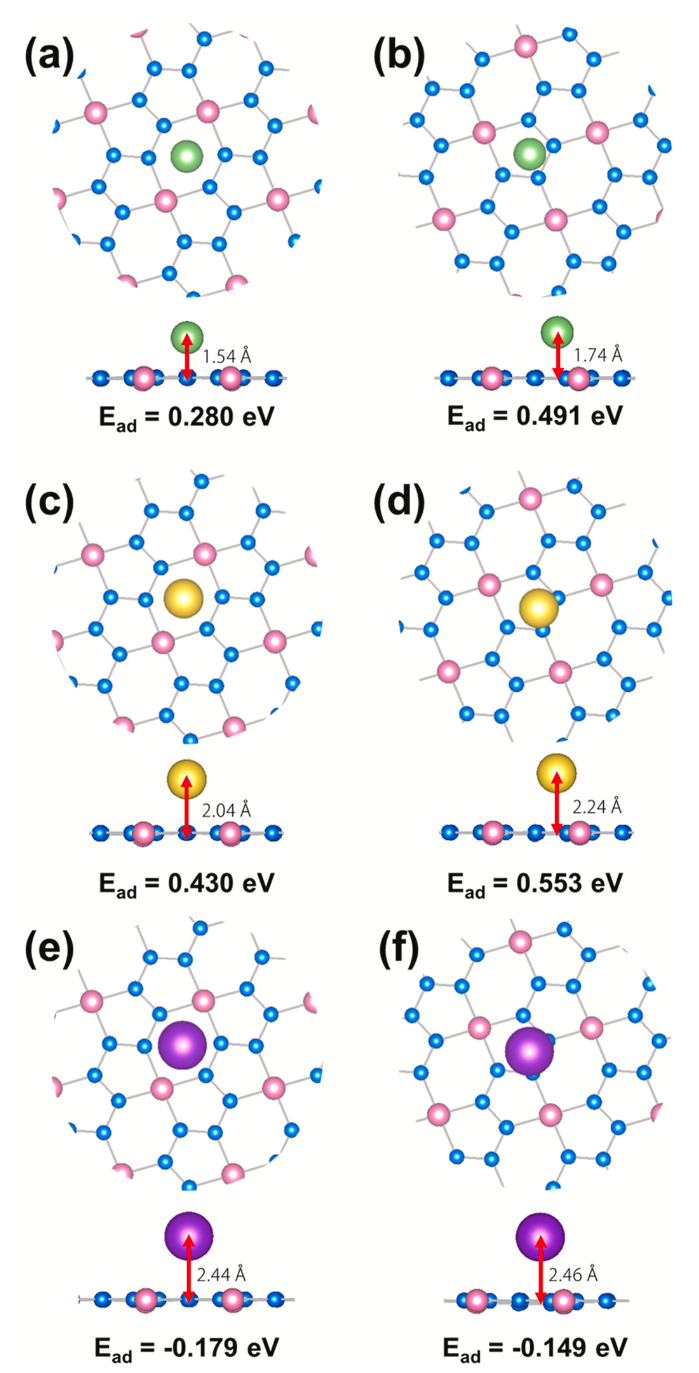
图2. BeN4单层吸附Li/Na/K后的优化结构的顶部和侧面图。Li吸附在BeN4单层的(a)H1位点和(b)H2位点。Na吸附在BeN4单层的(c)H1位点和(d)H2位点。K吸附在BeN4单层的(e)H1位点和(f)H2位点。吸附高度和吸附能量被标记在图中。
图3显示了K吸附在BeN4单层(a)H1位点和(b)H2位点上的差分电荷密度图,发现大量的电荷从K原子转移到BeN4单层,通过Bader电荷分析计算得到的电荷转移值分别为0.880 e/atom和0.881 e/atom(在H1和H2位点),此外K原子被转化为K离子并被紧紧地吸附在BeN4单层的表面。最后,对比了吸附K前后的BeN4单层的态密度(DOS),发现,所有这些DOS图在费米能级附近都有很高的电子密度,这表明BeN4单层在吸附K前后都有很好的导电性,这对于电极材料是至关重要的。
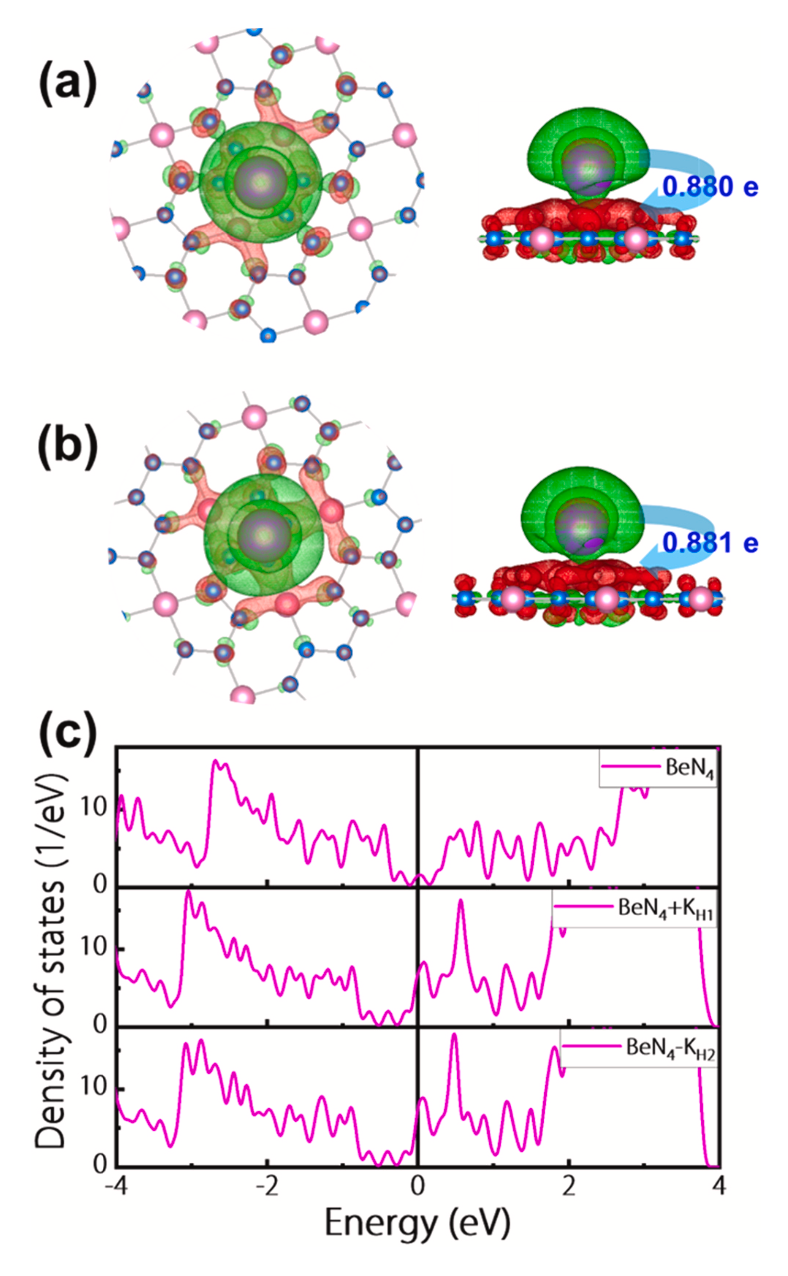
图3. K吸附在BeN4单层(a)H1位点和(b)H2位点上的差分电荷密度图。等表面值被设定为0.01 e Å-3。(c) 原始BeN4单层和BeN4单层吸附K后的态密度图(在H1和H2位点)。
图4显示了K离子在BeN4单层表面的两种可能的扩散路径,分别标记为Path1和Path2。其中,Path1和Path2代表K离子从H1位点沿不同方向扩散到邻近的另一个H1位点。此外,还显示了K离子在BeN4单层上的扩散障碍曲线,发现Path1是最快的K扩散路径,相应的最低扩散障碍是62 meV,这比许多已知的二维阳极材料的扩散势垒还要低。
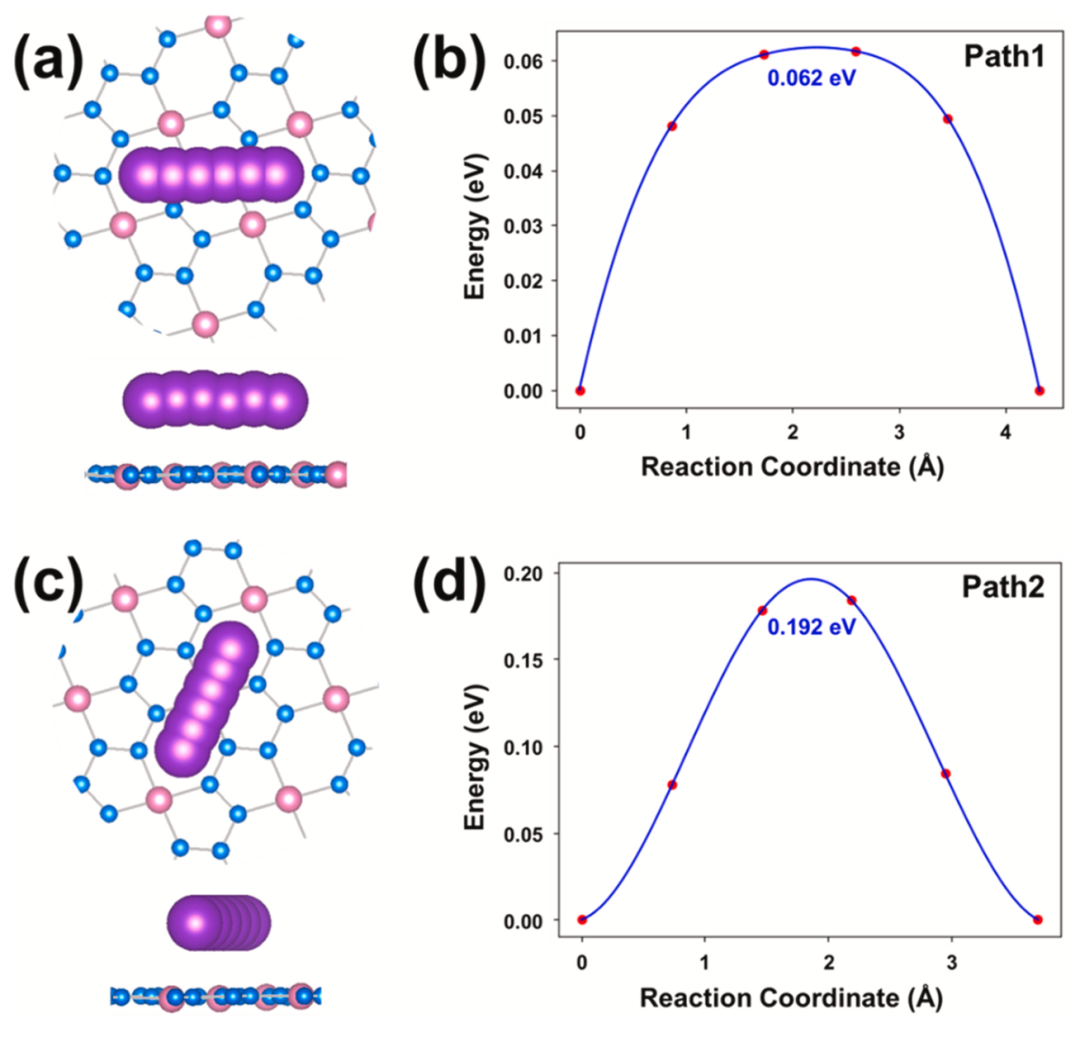
图4.(a)和(c)K离子在BeN4单层上的潜在扩散路径。(a)和(c)相应的K离子在BeN4单层上的扩散势垒曲线图。
图5显示了BeN4单层的对K的平均吸附能以及开路电压随吸附K的数量变化的曲线图,发现BeN4单层可以稳定吸附18个K,其对应的开路电压范围为0.084-0.179 V。其中,BeN4单层中不同K浓度的优化结构,相应的化学成分分别为BeN4K2/3、BeN4K4/3和BeN4K2也显示在图中。
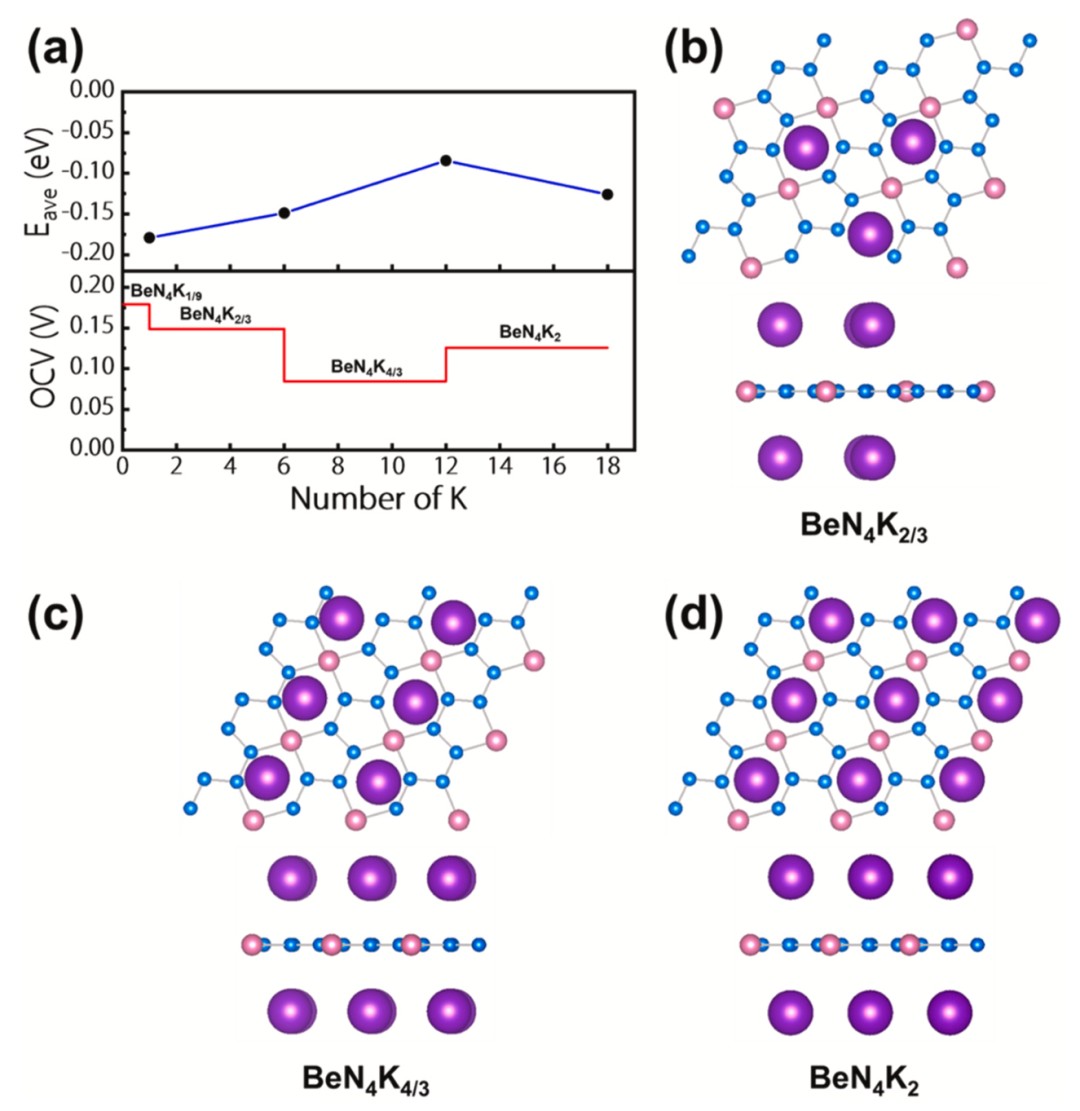
图5.(a)BeN4单层的对K的平均吸附能以及开路电压随吸附K的数量变化的曲线图。BeN4单层中不同K浓度的优化结构的俯视图和侧视图:(b)BeN4K2/3;(c)BeN4K4/3;(d)BeN4K2。
图6显示了BeN4K2结构在300 K和500 K下的AIMD模拟计算,发现BeN4K2的构象没有被破坏,只有轻微的变形,且其总能量只随时间只有轻微波动。所有这些结果表明BeN4单层在室温或者更高温度下插入钾后仍具有良好的热稳定性。
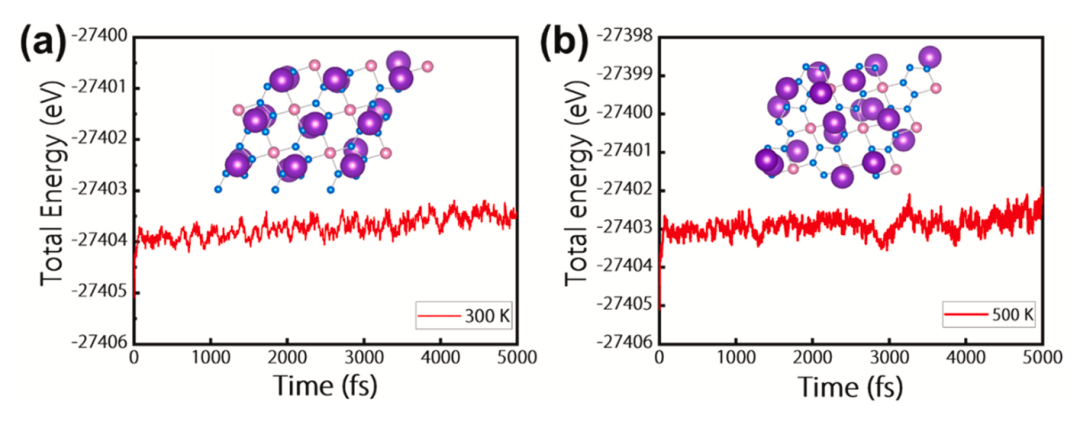
图6. BeN4K2结构在(a)300 K和(b)500 K下的AIMD模拟计算。
图7显示了BeN4单层与其他典型的钾离子电池二维阳极材料在扩散屏障和储存能力的对比,发现BeN4单层不仅具有很低的扩散势垒,且其对K的储存容量也高于绝大多数经典钾离子电池二维阳极材料。
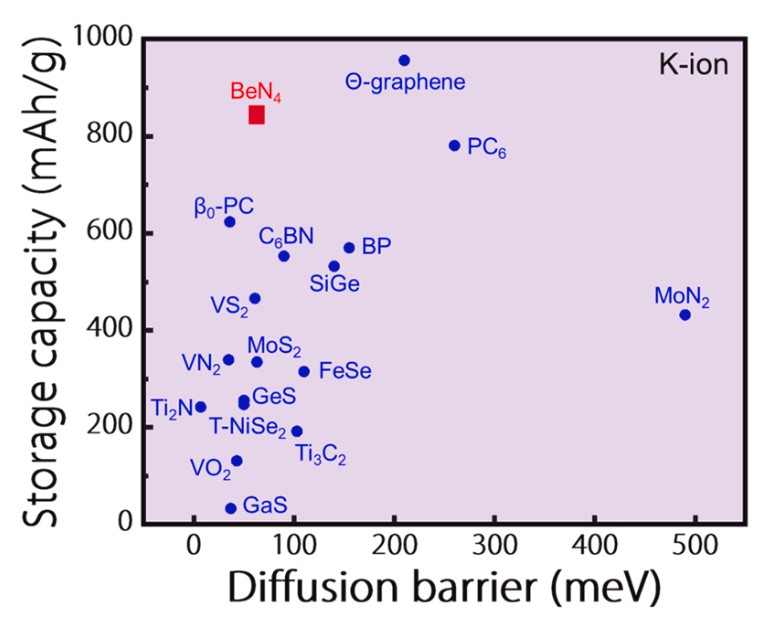
图7. 将BeN4单层与其他典型的钾离子电池二维阳极材料在扩散屏障和储存能力方面进行比较。
04 小结
最后,本文基于密度泛函理论,应用了鸿之微Devie Studio平台及DS-PAW软件工具,系统地研究了BeN4单层的晶体结构、稳定性和电子特性。考虑到其良好的物理和化学性能,进一步研究了BeN4单层作为MIBs阳极材料的可行性。其研究结果表明,只有钾可以稳定地吸附在BeN4单层的表面,并且具有Dirac锥的BeN4单层在吸附K前后都具有良好的导电性。更重要的是,BeN4单层具有相当低的扩散势垒和开路电压,但具有非常高的存储容量,这些都优于大多数PIBs的二维阳极材料。此外,BeN4单层还被证明具有长的循环寿命和非常好的循环稳定性。因此,这些结果表明,BeN4单层可以成为优异的钾离子电池狄拉克阳极材料。
审核编辑:刘清
-
锂离子电池的最基本知识2008-06-03 0
-
动力锂离子电池原材料2009-08-11 0
-
锂离子电池的设计2013-05-20 0
-
锂离子电池的基本组成及关键材料2013-07-03 0
-
有机化合物可作为锂离子电池正极材料2015-11-17 0
-
关于锂电池电极材料SEM测试、氩离子截面解剖电极片2017-07-07 0
-
锂离子电池简介2020-11-03 0
-
锂电材料截面制样-氩离子抛光CP离子研磨 金鉴实验室分享(上)2020-12-16 0
-
如何选择动力型锂离子电池的正极材料?2021-05-12 0
-
锂离子电池隔膜材料2009-10-22 745
-
碳纳米管来制造硅阳极锂离子电池,开拓锂离子电池电极材料的使用2020-04-08 2471
-
铌作为电池阳极的金属有何特别之处2022-08-04 1829
-
锌离子电池下一代先进阳极的的协同保护策略2022-11-14 1303
-
锂电池和锂离子电池的区别?锂离子电池充电模式2023-10-24 1290
全部0条评论

快来发表一下你的评论吧 !
