

上海交大JACS:单原子催化,非晶态载体更具优势!
描述
通讯作者:占光明、么艳彩、张礼知
通讯单位:上海交通大学
文章链接:https://pubs.acs.org/doi/10.1021/jacs.3c13834
01
导读
氯气(Cl2)在有机合成、家用漂白剂和水处理中有着广泛的应用。工业生产Cl2需要在饱和氯化钠以及强酸性溶液条件下(pH值约2)下进行电解,导致阳极氯析出反应(CER)需要使用到大量DSA(负载于Ti基底的Ru/Ir氧化物)来加速电化学反应。为了可持续发展,减少贵金属用量和提高性能是CER电极发展的迫切要求。
02
成果简介
作者报道了非晶态钛氧化物载体能够有效调节Ir单原子的配位环境,从而实现钛阳极高效持久的析氯反应。实验和理论结果表明,在非晶钛氧化物和结晶钛氧化物上分别形成了四配位Ir1O4和六配位Ir1O4结构。有趣的是Ir1O4位点表现出优越的CER性能,其质量活性分别是IIr1O4和DSA的10倍和500倍。此外,Ir1O4阳极表现出优异的200小时耐久性,远远超过Ir1O4阳极(2小时)。机理研究表明,Ir1O4中的不饱和Ir位点是CER的活性中心。Ir1O4的无定形结构和受限的水解离协同阻止了O在Ti基底上的渗透,有助于其长期的CER稳定性。该成果以“Engineering the Coordination Environment of Ir Single Atoms with Surface Titanium Oxide Amorphization for Superior Chlorine Evolution Reaction”为题发表在国际顶级期刊Journal of the American Chemical Society上。
03
研究亮点
1,作者提出了钛氧化物载体的结晶性能够调节单原子Ir的配位结构:在非晶钛氧化物和结晶钛氧化物上分别形成了四配位Ir1O4和六配位Ir1O6结构。单原子Ir的结构差异将影响物种的吸附。
2,Ir1O4显示出优异的CER性能,过电位仅为71.8 mV@10 mA cm-2,且具有200小时耐久性。
3,实验与模拟结果显示,氧化层的非晶化结构以及缓慢的水解离有效阻止了氧渗透到Ti基底内部,提高了Ti的抗钝化能力。
04
研究内容
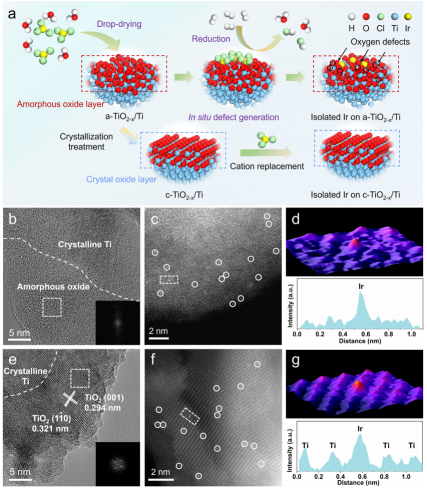
图1 合成及电镜表征:(a)在a-TiO2-x/Ti和c-TiO2-x/Ti上合成单原子Ir的示意图;(b,e)a-TiO2-x/Ti和c-TiO2-x/Ti的HRTEM图像;(c,f)a-TiO2-x/Ti和c-TiO2-x/Ti的AC HAADF-STEM图像;(d,g)分别沿着(c)和(f)中矩形标记的区域对应的三维AOGF映射和线强度分析。
通常,天然形成的钛氧化物的结晶度较差,其内部有晶态Ti,表面有非晶态钛氧化物层,边界清晰(记为a-TiO2-x/Ti)。通过对a-TiO2-x/Ti进行高温处理和慢冷相结合的结晶处理,制备出相应的晶体(c-TiO2-x/Ti),使其逐渐由非晶态转变为晶态。
接着,作者利用缺陷工程策略和金属-载体相互作用,将单原子Ir固定在不同晶相的钛氧化物上。通过HAADF-STEM可以看到,Ir分别分散在a-TiO2-x/Ti(图1b、c)和c-TiO2-x/Ti表面(图1e、f)。经三维原子重叠高斯函数拟合(3D AOGF),如图1d、g所示,其显示Ir呈现高度分散的孤立结构,与线强度分析一致。
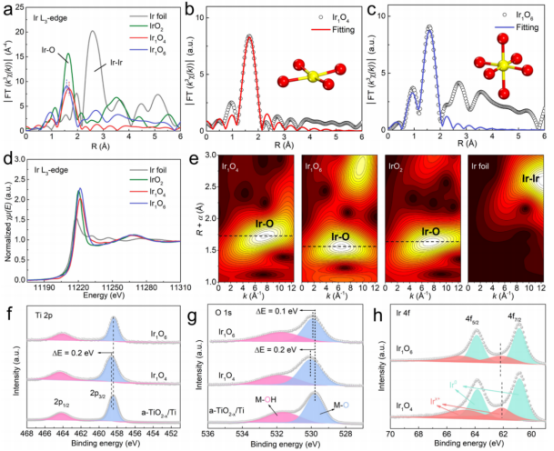
图2 结构表征:(a)Ir的L3边缘FT-EXAFS谱图;(b,c)Ir的L3边缘FT-EXAFS拟合曲线;(d)Ir的L3边缘XANES谱图;(e)小波变换光谱;(f-h)Ti 2p、O 1s、Ir 4f的XPS谱图。
接着,利用XAFS和XPS检测了单原子Ir的局部配位环境和电子态。两个样品的FT-EXAFS谱图均显示一个主峰,对应Ir-O配位,而没有出现Ir-Ir配位,表明Ir呈原子分散结构(图2a)。通过拟合EXAFS光谱(图2b、c),结果表明,在a-TiO2-x/Ti上,Ir仅与4个相邻的O配位(记为Ir1O4),表明Ir具有高度配位不饱和的特征;而负载于c-TiO2-x/Ti上的Ti与6个相邻的O配位(记Ir1O6。因此,Ir1O4中的Ir-O散射路径(~1.67 Å)比Ir1O6的Ir-O散射路径(~1.58 Å)更大,这是由于在非晶基底上,Ir-O原子间距被拉伸所致;图2d的小波变换也证实了这一结果(Ir1O4具有较高的R和k值)。
图2e的Ir的L3边缘XANES谱图显示,与Ir1O6相比,Ir1O4中的Ir的L3边缘的峰强度较低,即由载体非晶化引起Ir的平均价态较低。与a-TiO2-x/Ti基底相比,Ir1O4中Ti 2p3/2的峰偏移到更高的结合能处,表明在Ir沉积后,电子可能从Ti转移到单原子Ir。结合Ir1O4中明显正移的金属-氧峰(M-O)(图2g),可以推断电子转移发生在Ti-O-Ir上。相反,对于Ir1O6,Ti 2p3/2和M-O的峰位移较小,表明其Ti-O-Ir单元的电子转移较弱。这一推测也得到了Ir1O4中Ir 4f7/2具有较低的结合能的支持(图2h),表明电子从Ti基底转移到单原子Ir上。因此,Ti基底的表面氧化层的结晶度将强烈影响单原子Ir的局部配位环境与电子结构(配位饱和的Ir1-O6结构可能难以吸附Cl物种),从而提供明显不同的CER活性。
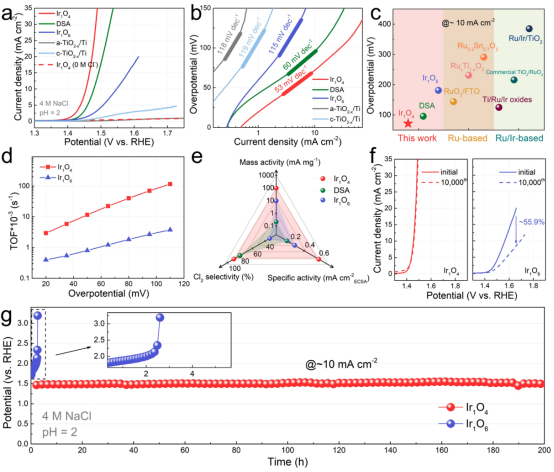
图3 CER性能:(a)CER极化曲线;(b)Tafel曲线;(c)不同催化剂的过电位比较;(d)Ir1O4和Ir1O6的TOF数值比较;(e)Ir1O4、Ir1O6和DSA的性能参数比较;(f)循环1万次后的极化曲线;(g)在10 mA cm-2下的计时电位曲线。
在pH=2的4 M NaCl中评估了Ir1O4、Ir1O6的CER性能,并将其与商业DSA、a-TiO2-x/Ti和c-TiO2-x/Ti进行了比较。图3a的LSV曲线显示,Ir1O4可以在更低的过电位下提供更高的电流密度。例如,在10 mA cm-2下,Ir1O4的过电位仅为71.8 mV,且Tafel斜率为53 mV dec-1,这一结果远优于Ir1O6(181.4 mV和115 mV dec-1), DSA (95.8 mV和60 mV dec-1),以及大多数的Ru和Ru/Ir基CER电催化剂(图3b、c)。
图3d进一步比较了Ir1O4、Ir1O6的转换频率(TOF)。例如,Ir1O4在过电位为110 mV时的TOF为0.12 s-1,比Ir1O6的TOF高31倍。同时,由ECSA归一化的比活性也揭示了Ir1O4具有更高的本征活性(图3e),优于商业DSA与Ir1O6。
在没有NaCl的情况下,Ir1O4几乎不产生法拉第电流(图3a),表明其OER活性极差。在电解质中加入NaCl后,Ir1O4的Cl2选择性达到90.0%,与Ir1O6(32.5%)和DSA(80.1%)形成鲜明对比(图3e),证实了其优异的Cl2选择性。更重要的是,Ir1O4表现出优异的耐久性,在循环1万次前后的极化曲线、以及200 h的长时间运行中都没有明显的活性衰减(图3f、g)。

图4 CER机制:(a)Ir1O4和Ir1O6中单原子Ir的局部配位构型;(b)差分电荷密度图;(c)CER过程的自由能图;(d,e)CER条件下的原位拉曼光谱;(f)吸附氯原子后,Ir1O4和Ir1O6的DOS分布;(g)反应后收集Ir1O4和Ir1O6,进行H2-TPR处理;(h)在m/z=36时,收集到的H2-TPR谱线。
作者采用DFT计算来理解Ir1O4优异的CER活性和活性位点。根据EXAFS拟合结果,构建了Ir1O4和Ir1O6的合理模型,如图4a所示。差分电荷密度图显示单原子Ir和基底之间存在电子相互作用,Ir1O4的Bader电荷转移(+1.15e)低于Ir1O6的Bader电荷转移(+1.40e),即更多的电子从a-TiO2-x/Ti转移到单原子Ir上。图4c显示了CER过程的自由能,Ir1O4上亲电不饱和Ir位点的Cl吸附能为-0.1 eV,比Ir1O6上配位O位点的Cl吸附能(+0.31 eV)更接近于0 eV,有利于Cl的吸附。
因此,暴露的单原子Ir是Ir1O4的Cl吸附位点,这也被原位拉曼光谱证实(图4d):随着反应电位的增加,在500 cm-1处出现了一个越来越强的Ir-Cl键拉曼信号。相比之下,在饱和配位的Ir1O6催化剂上,只在791 cm-1处观察到一个较弱的峰(O-Cl键)(图4e)。空间位阻效应增加了Cl在饱和Ir配位位点上的吸附难度,但有利于其在上配位O原子上的吸附。
通过态密度(DOS)进一步比较不同吸附位点与Cl之间的相互作用(图4f)。Ir1O4中Ir 5d和Cl 3p轨道的重叠比Ir1O6中O 2p和Cl 3p轨道的重叠要大,表明Cl与不饱和Ir位点的相互作用强于与饱和Ir的顶配位O的相互作用。通过H2程序升温还原(H2-TPR)进一步验证了Cl物种在Ir1O4和Ir1O6上的吸附强度,发现Ir1O4中Cl解吸峰的温度高于Ir1O6(图4g、h),证实了Ir1O4对Cl的吸附更强,这与理论计算结果和原位拉曼光谱中更强的吸附信号相吻合。上述分析可以简单理解:CER催化活性的巨大差异源于Ir1O6上Cl的吸附位点从顶部配位的O到Ir1O4中不饱和Ir位点的变化,从而影响了与Cl的结合强度。
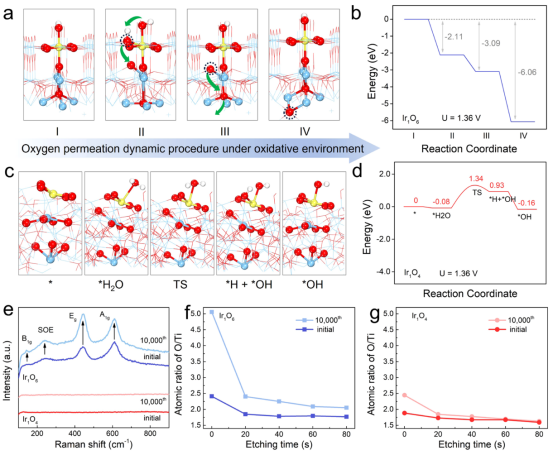
图5 失活机理研究:(a)氧化环境下Ir1O6可能的氧渗透动态过程;(b)氧渗透动力学过程的相应自由能图;(c)Ir1O4在水吸附和随后解离成羟基过程中的局部结构构型和过渡态;(d)水吸附和解离的自由能图;(e)Ir1O4和Ir1O6在10000圈循环前后的拉曼光谱;(f,g)不同蚀刻时间后O/Ti的原子比。
通过理论模拟分析了结晶钛氧化物的钝化过程(图5a、b)。由于整个CER过程发生在酸性溶液中,处于初始状态的Ir1O6表面以表面吸附羟基(*OHsurf)的形式暴露出来(阶段I)。在施加电位(1.36 V)下,表面配位饱和的Ir1O6会自发吸附另一个OH,吸附能为-2.11 eV,达到过饱和状态(阶段I~II)。初始态的*OHsurf由于位阻的排斥力进入亚表面,自发脱氢形成亚表面氧(Osub,阶段II~III),新的*OHsurf将占据原来*OHsurf的位点。在氧化层和金属层之间氧浓度差的驱动下,新形成的Osub可以沿着Ti-O-Ti自发地渗透到氧化层和金属层的界面中(阶段III~IV),从而使氧化层变厚,导致电子转移阻抗的急剧增加。总的来说,在Ir1O6上,OH的过饱和吸附(阶段I到II)、OH的脱氢渗透(阶段II到III)以及随后Osub的深度渗透(阶段III到IV)都是自发过程。
在氧渗透过程中,氧化层和金属层之间的氧浓度差是动力,而规则且排列紧密的Ti-O-Ti框架通过提供轴向通道促进了这一过程。对于Ir1O4,由于水解离存在较大的势垒(1.42 eV),阻止了额外的OH吸附(图5c、d)。因此,Ir1O4催化剂上的氧渗透和Ti基底的钝化被抑制。
反应前后电极的拉曼光谱证实了Ir1O6上氧化层的增厚(图5e),反应后在143、239、443和610 cm-1处的拉曼信号明显增加,对应金红石型TiO2结构,而Ir1O4没有观察到这种情况,这与其无定形氧化层的无序特性有关。
通过Ar离子蚀刻的XPS光谱进一步验证了这种抗钝化机制,定性分析了电极表面和亚表面上O/Ti原子的比例(图5f、g)。新鲜的Ir1O4和Ir1O6电极表面的O/Ti原子比很低,小于2。然而,反应后Ir1O6的表面O的比例明显增加,这是由于氧的吸附能力过大。随着刻蚀时间的增加,O/Ti比值甚至高于初始样品(>2),证实了氧渗透到电极中,使电极钝化(图5f)。经过1万次循环反应后,Ir1O4的表面O比例仅略有增加,而亚表层的O/Ti比例与初始样品非常接近,有力地验证了氧化层非晶化的抗钝化能力(图5g)。上述分析可以简单理解:对于Ir1O4,无定形氧化层结构和较高的水解离势垒,有效阻止了氧渗透到Ti基底内部,抑制了Ti钝化,使Ir1O4显示出优异的CER稳定性。
05
总结与展望
综上所述,本文证明了表面钛氧化物非晶化是一种有效的策略,可用于调节单原子Ir的配位环境,以实现高效和稳定的CER。所制备的Ir1O4电极在10 mA cm-2下过电位仅为71.8 mV,质量活性为95 mA mgIr-1,远远优于Ir1O6和DSA。原位拉曼光谱和理论模拟表明,CER催化活性的巨大差异源于Ir1O6上Cl的吸附位点从顶部配位的O到Ir1O4中不饱和Ir位点的变化,从而影响了与Cl的结合强度。更重要的是,钛氧化物的无定形结构和缓慢的水解离阻止了氧渗透到Ti基底内部,抑制了Ti钝化,使Ir1O4阳极具有强大的稳定性。这些发现强调了表面氧化物结晶度对钛基阳极活性和稳定性的影响,并对如何调节单原子配位环境提供了见解,以获得更好的催化性能。
审核编辑:刘清
-
嵌入式开发课件(PDF)[上海交大]2012-08-14 0
-
上海交大控制理论讲义2012-08-20 0
-
载体催化元件电路图2009-06-08 980
-
非晶态结构2009-08-06 2405
-
Silvaco公司与上海交大成立半导体与显示器件教学联合实验室2019-07-26 3492
-
宁德时代与上海交大合作,双方将共建清洁能源技术联合研究中心2020-10-16 1936
-
上海交大黑科技“小叶子”24小时监控体温 、低功耗蓝牙无线连接2020-11-04 1711
-
高活性生物质碳负载Fe/Pt单原子双功能催化剂开发2021-02-12 2488
-
联想集团捐建给上海交大高性能计算中心正式揭牌启用2021-12-16 1745
-
非晶态纳米棒用于高电流密度下电催化中性水分解2022-12-30 1160
-
双原子催化剂综述:适用于能源和环境催化的双原子催化剂2023-07-17 4316
-
云天励飞与上海交大国际与公共事务学院合作推动人工智能产业快速发展2023-10-31 631
-
季丰电子荣膺2023上海交大“未来领军”企业2023-11-25 375
-
上海交大与云天励飞签署战略合作协议,寻找AI时代的Killer App2024-04-22 266
全部0条评论

快来发表一下你的评论吧 !
