

基于边合成边测序技术的数据质量控制
电子说
描述
基于边合成边测序(Sequencing By Synthesis,SBS)技术,Illumina HiSeq2500高通量测序平台对cDNA文库进行测序,能够产出大量的高质量Reads,测序平台产出的这些Reads或碱基称为原始数据(Raw Data),其大部分碱基质量打分能达到或超过Q30。Raw Data通常以FASTQ格式提供,每个测序样品的Raw Data包括两个FASTQ文件,分别包含所有cDNA片段两端测定的Reads。
FASTQ格式文件示意图如下:

FASTQ格式文件示意图
注:FASTQ文件中通常每4行对应一个序列单元:第一行以@开头,后面接着序列标识(ID)以及其它可选的描述信息;第二行为碱基序列,即Reads;第三行以“+”开头,后面接着可选的描述信息;第四行为Reads每个碱基对应的质量打分编码,长度必须和Reads的序列长度相同。
测序碱基质量值
碱基质量值(Quality Score或Q-score)是碱基识别(Base Calling)出错的概率的整数映射。通常使用的Phred碱基质量值公式为: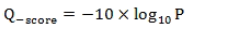
公式中,P为碱基识别出错的概率。下表给出了碱基质量值与碱基识别出错的概率的对应关系:
表1 碱基质量值与碱基识别出错的概率的对应关系表

碱基质量值越高表明碱基识别越可靠,碱基测错的可能性越小。比如,对于碱基质量值为Q20的碱基识别,100个碱基中有1个会识别出错;对于碱基质量值为Q30的碱基识别,1,000个碱基中有1个会识别出错;Q40表示10,000个碱基中才有1个会识别出错。
以测序循环为单位,对单个样品所有Reads平行测序的碱基质量值做分布图,可以查看单个样品各个测序循环及整体的测序质量。
碱基质量值分布图
注:横坐标为测序碱基在Reads上的位置,纵坐标为碱基质量值。颜色深浅表示碱基比重,颜色越深,说明该位置测定的碱基中为对应质量值的碱基所占的比重越大,反之亦然。
测序质量控制
FASTQ文件中测序Reads需要与指定的参考基因组进行序列比对,定位cDNA片段在基因组或基因上的位置。在序列比对之前,首先需要确保这些Reads有足够高的质量,以保证后续分析的准确。测序质量控制方式如下:
(1) 去除测序接头以及引物序列;
(2) 过滤低质量值数据,确保数据质量。
经过上述一系列的质量控制之后得到的高质量Reads或碱基,称为Clean Data。Clean Data同样以FASTQ格式提供。
测序数据产出统计
某项目各样品数据产出统计见下表:
表2 样品测序数据评估统计表
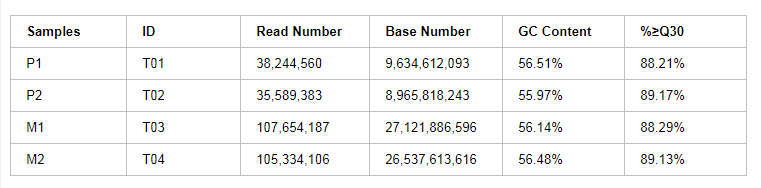
注:Samples:样品信息单样品名称;ID:样品编号;Read Number:Clean Data中pair-end Reads(双末端测序)总数;Base Number:Clean Data总碱基数;GC Content:Clean Data GC含量,即Clean Data中G和C两种碱基占总碱基的百分比;%≥Q30:Clean Data质量值大于或等于30的碱基所占的百分比。
转录组数据与参考基因组序列比对
获得Clean Reads后,将其与参考基因组进行序列比对,获取在参考基因组或基因上的位置信息,以及测序样品特有的序列特征信息。
TopHat2是一个高效的序列比对软件。它以高通量Reads比对软件Bowtie为基础,将转录组测序Reads比对到基因组上,然后通过分析比对结果识别外显子之间的剪接点(Splicing Junction)。这不仅为可变剪接分析提供了数据基础,还能够使更多的Reads比对到参考基因组,提高了测序数据的利用率。
转录组测序数据中,只有比对到参考基因组上的数据才能用于后续分析。因此,将比对到指定的参考基因组上的Reads称为Mapped Reads,对应的数据称为Mapped Data。
比对效率统计
比对效率指Mapped Reads占Clean Reads的百分比,是转录组数据利用率的最直接体现。比对效率除了受数据测序质量影响外,还与指定的参考基因组组装的优劣、参考基因组与测序样品的生物学分类关系远近(亚种)有关。因此,通过比对效率,可以评估所选参考基因组组装是否能满足信息分析的需求,及后期数据分析的可靠性。
各样品测序数据与所选参考基因组的序列比对结果统计见下表:
表3 Clean Data与参考基因组比对结果统计表
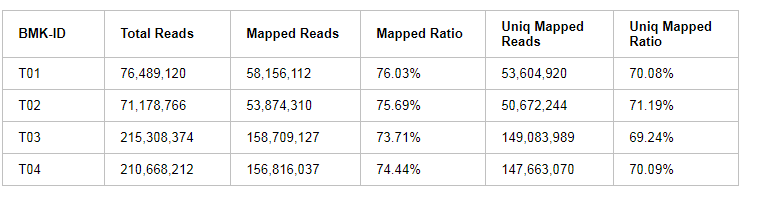
注:ID:样品编号;Total Reads:Clean Reads数目,按单端计;Mapped Reads:比对到参考基因组上的Reads数目;Mapped Ratio:比对到参考基因组上的Reads在Clean Reads中占的百分比;Uniq Mapped Reads:比对到参考基因组唯一位置的Reads数目;Uniq Mapped Ratio:比对到参考基因组唯一位置的Reads在Clean Reads中占的百分比。
比对结果作图
将比对到不同染色体上Reads进行位置分布统计,绘制Mapped Reads在所选参考基因组上的覆盖深度分布图。
样品T01的Mapped Reads在参考基因组部分染色体上的覆盖深度分布图如下:
Mapped Reads在参考基因组上的位置及覆盖深度分布图
注:横坐标为染色体位置;纵坐标为覆盖深度以2为底的对数值,以10kb作为区间单位长度,划分染色体成多个小窗口(Window),统计落在各个窗口内的Mapped Reads作为其覆盖深度。
理论上,来自成熟mRNA的Reads应该比对到外显子区。但是,由于以下原因一部分Reads会比对到内含子区和基因间区:
(1) 样品提取时将含有Ploy(A)尾而内含子没有切除完全的mRNA(即mRNA前体)提出,使得来自内含子片段的Reads比对到了内含子区;
(2) 基因组注释错误,原来为外显子的区域注释成了内含子区,或者相反;
(3) 基因组注释水平低,对于使用转录组测序数据进行的基因组注释,由于转录组测序不能遍历所有的时间和空间点,使得用于注释的转录组测序数据中不表达或低表达的基因刚好在该项目的样品中检测到较高丰度时,来自这类基因的Reads就比对到了被注释的基因间区,这也是新基因和新转录本发掘的基础之一;
(4) 测序样品与参考基因组存在差异,比如测序样品中突变形成新的转录组起始位点形成样品特有的新基因,或者剪接位点差异形成新的转录本,这也是新转录本发掘的基础之一。
统计Mapped Reads在指定的参考基因组不同区域(外显子、内含子和基因间区)的数目,绘制基因组不同区域上各样品Mapped Reads的分布直方图,如下:
基因组不同区域Reads分布直方图
注:图中每个直方柱表示一个样品,粉色区域为外显子区、绿色区域为基因间区、蓝色区域为内含子区,区域的高度表示比对到该区域的Mapped Reads在所有Mapped Reads中所占的百分比。
编辑:hfy
-
轮边驱动电机专利技术发展2025-06-10 467
-
电源设计中的原边反馈控制和副边反馈控制方案分析2022-11-10 10863
-
基于边合成边测序(SBS)原理的测序芯片和测序试剂盒2020-06-30 9734
-
基于边分割的社交网络敏感边保护技术2017-12-26 867
-
基于模糊综合评价的接边质量评价2017-12-13 869
-
基于AC/DC控制芯片的原边反馈技术2012-11-08 4413
-
原边反馈AC/DC控制芯片中的关键技术2012-03-08 4388
-
有源箝位变压器的高边或低边调整技术2011-10-14 871
-
SMT车间管理与质量控制技术(续完2010-04-24 2101
全部0条评论

快来发表一下你的评论吧 !
