

AlphaFold有望改变生物学
描述
AlphaFold有望改变生物学。
近日,谷歌AI团队DeepMind所研究的 AlphaFold 算法在生物学领域取得了重要突破:通过蛋白质的氨基酸序列高精度地确定其3D结构。
具体而言,是DeepMind的第二代AlphaFold 在国际蛋白质结构预测竞赛(CASP)上击败了其余的参会选手,能够精确地基于氨基酸序列,预测蛋白质的3D结构。其准确性可以与使用冷冻电子显微镜(CryoEM)、核磁共振或 X 射线晶体学等实验技术解析的3D结构相媲美。
前基因泰克(Genentech)首席执行官Arthur D. Levinson博士称这一成就为“划时代的进步”(once in a generation advance)。
从CASP14,这项权威的蛋白质结构预测竞赛结果中,可以看到AlphaFold2排名第一(总分远高于第二),排名前六位的分别是David Barke lab的两个工具(第二、第三位)、密歇根州立大学的 Michael Feig和密歇根大学Yang Zhang排名在第四位、第五位,而腾讯AI lab 的tFold_human 排名第六。
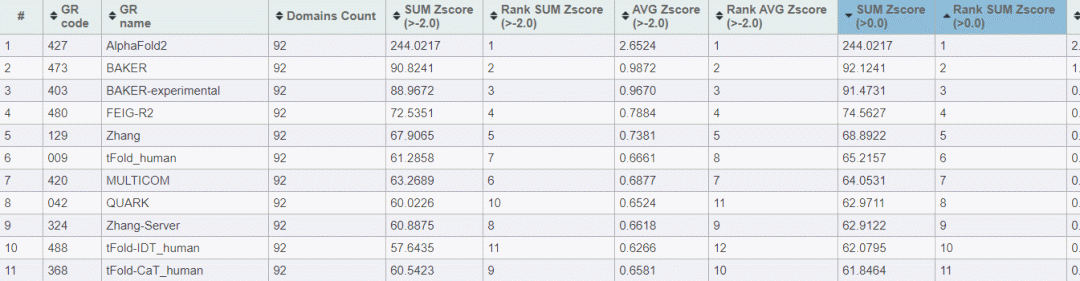
这说明,在某种程度上,在对蛋白质结构和折叠的预测这件事情上,人类已非望尘莫及。
第二代AlphaFold做了什么?
在CASP这项比赛中,DeepMind开发AlphaFold2用的数据是:数据库里的超过17万种蛋白序列与结构,以及其他一些大型数据库里的数据。
评估蛋白结构预测准确度的指标叫做GDT,也就是评估预测结构里的氨基酸位置,和实际的氨基酸位置差多少。差得越少,得分越高。
GDT的分值在0-100之间。2006-2016年间,这个数字最高在40左右。2018年,上一代的 AlphaFold得分一下子突破了50。而这次的新一代 AlphaFold,在蛋白结构预测大赛里的中位得分超过了92.4。
不仅仅远超今年的竞争对手,相比于2018年版的AlphaFold第一代,AlphaFold2的表现也如脱胎换骨一般。
更直观一些。拿它预测的结构与实际结构对比,可以看到基本完全吻合(下图,绿色是实验得到的实际结构,蓝色是计算预测结构)。
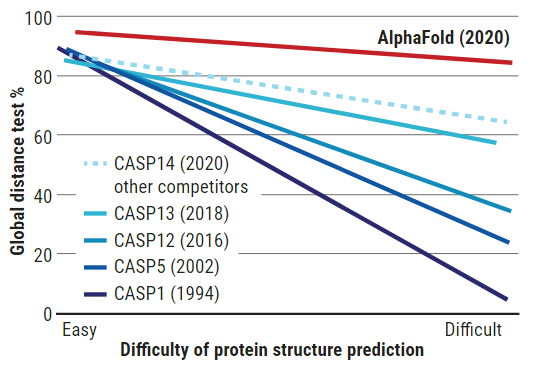
在两年一次的蛋白质结构预测关键评估(CASP)竞赛中,AlphaFold今年击败了所有其他小组,并在准确率方面与实验结果相匹配。随着预测难度增加,AlphaFold的准确率保持在稳定的高水平,表现远超其它团队以及往届竞赛的水平。
但是对此结果,也有不少人存在疑问,主要在准确性。首先,17万的数据应该远远不够,准确性存疑;再者,蛋白质结构折叠问题太过高深,如果真能够破解,DeepMind很快就能获得诺贝尔奖。
因此,在惊叹AlphaFold取得成果的同时,我们还需要冷静等待生物学家的实验验证。
除了解决的问题值得我们关注,其实研究方法也更有思考价值。
毕竟,AlphaFold的结构预测与X射线晶体学或低温电子显微镜(cryo-EM)等标准实验方法来预测并无区别,只是后者更加费力、更加昂贵。科学家表示,AlphaFold的出现也许并不会完全替代这些实验方法,但确实为人们提供了研究生物的新方式。
蛋白质结构:生物学五十年来的挑战
蛋白质是生命的基础,与细胞组成内容紧密相关。而蛋白质的功能取决于其3D结构。
一直以来,生物学家投入实验,致力于寻找生命的奥秘,即:氨基酸序列(蛋白质的组成部分)是如何绘制出最终的形状。
在过去,人们一直是通过实验室来了解蛋白质的结构。比如,使用X射线束照射结晶的蛋白质,并将衍射光转化为蛋白质原子坐标,由此掌握蛋白质的第一个完整结构。
除了实验,随着计算机的发展,该项技术在上世纪末便已用于预测蛋白质的结构,但效果并不理想。
计算机技术用于蛋白质结构预测的困境,直到AlphaFold在2018年现身于CASP,才让科学家重新燃起了信心与希望。
AlphaFold的第一次迭代将深度学习应用于结构和遗传数据,以预测蛋白质中氨基酸对之间的距离。根据AlphaFold的主要负责人之一John Jumper介绍,在第二个步骤中,虽然没有使用AI技术,但AlphaFold使用结构与遗传数据得出了蛋白质的外观模型,与之前的研究达成一致。
但第一次迭代存在缺陷。因此,该团队开发了一个AI网络。该网络结合了有关确定蛋白质如何折叠的物理和几何约束的信息。他们设立了一项艰巨的目标:该网络能够预测目标蛋白质序列的最终结构,而不仅是预测氨基酸之间的关系。
惊人的准确性
CASP的进行历时数月。
1994年,Moult及其同事发起了CASP,每两年召开一次。比赛团队获得约100种蛋白质的氨基酸序列,其结构未知。一些小组计算每个序列的结构,而另一些小组则通过实验确定它。然后,组织者将计算预测结果与实验室结果进行比较,并为预测结果提供全球距离测试(GDT)分数。
研究小组有数周的时间来提交其结构预测。然后,一组独立的科学家使用度量标准来评估预测的蛋白质与实验确定的结构的相似程度,以此评估各个研究小组的预测结果。研究小组的名称为匿名。
在今年的比赛中,AlphaFold的称号是“427组”。其预测的许多条目具有惊人的准确性,将近三分之二的预测与实验所得的结构相当。
针对各种目标蛋白,AlphaFold的GDT中位数得分为92.4。在难度中等的蛋白质上,其他团队的最佳表现通常在预测准确度上得分75(满分100),而在AlphaFold则得分大约90,中位数为87,比下一个最佳预测高25分。
AlphaFold甚至擅长解决楔入细胞膜中的蛋白质结构,这是许多人类疾病的核心,但众所周知,这个问题用X射线晶体学都很难解决。医学研究理事会分子生物学实验室的结构生物学家Venki Ramakrishnan称该结果“在蛋白质折叠问题上取得了惊人的进步。”
Moult介绍,90分以上得分的预测结果与实验方法相当。
不过,AlphaFold并非完美胜任所有的预测。在一种由52个小重复片段组成、组装时会扭曲彼此位置的蛋白质上,AlphaFold的预测与实验结果之间存在部分差异。
CASP的负责人Moult介绍,无法确定是因为AlphaFold的预测误差,还是因为实验的伪像。
此外,AlphaFold的预测与通过核磁共振成像技术确定的实验结构的匹配度差,可能是因为AlphaFold将原始数据转换为模型的方法需要改进。
再如,AlphaFold的网络尝试为蛋白质复合物或组中的单个结构建模,因此,与其他蛋白质的相互作用扭曲了它们的形状。
应用
AlphaFold的预测有助于确定Lupas实验室多年来试图破解的细菌蛋白质的结构
Lupas的研究小组以前曾收集过原始的X射线衍射数据,但要将这些类似Rorschach的图案转换为结构,则需要了解蛋白质的结构信息。Lupas介绍:“在我们花了十年时间尝试一切之后,427组的模型在半小时内为我们提供了结构。”
DeepMind的联合创始人兼首席执行官Demis Hassabis介绍,AlphaFold可能需要几天的时间才能预测出蛋白质的结构,包括对蛋白质不同区域的可靠性估计。但AlphaFold将开放给科学家使用。
Hassabis认为,AlphaFold有望应用于药物发现和蛋白质设计。
借助AlphaFold,药物设计人员能够快速确定危险的新病原体(如SARS-CoV-2)中各种蛋白质的结构,这是寻找分子以阻止疾病产生的关键步骤。
加州大学伯克利分校的分子神经生物学家Stephen Brohawn说,DeepMind对一种叫做Orf3a的蛋白质的预测最终与后来通过cryo-EM确定的蛋白质非常相似。
AlphaFold的出现,可能意味着获得良好的蛋白质结构,不再局限于实验室,而只需要低质量的、易于收集的实验数据。比方说,类似蛋白质的进化分析等方向之所以能蓬勃发展,便是因为有大量的基因组数据能够被转化为结构。
科学家评论,AlphaFold能够帮助人类了解生命基因组中数千种未溶解蛋白质的功能,了解人与人之间因为疾病而产生的基因变异。
AlphaFold的出现也改写了外界对DeepMind的印象。此前,大家知道DeepMind,一般是因为该团队使用AI来玩游戏,比如AlphaGo。但如今,AlphaFold涉足生物学领域,例如进行蛋白质结构预测,DeepMind也因此向外界传达了自己的另一种声音:
不仅能玩围棋,还能使用AI帮助生命科学的长远发展。
责任编辑:lq
-
微流控技术的生物学应用2024-12-01 1118
-
合成生物是什么-微流控芯片技术在合成生物学的应用前景2024-05-28 2086
-
探索生物学中的电泳2022-10-10 5579
-
SnapGene记录你的日常分子生物学程序2022-08-30 1695
-
改变生物学研究进程:AI模型打开生命信息密码2021-12-16 4288
-
基于合成生物学技术生产的产品又有哪些优势呢?2021-06-22 3981
-
挽救生命的合成生物学解决方案2021-02-19 2020
-
合成生物学的发展与面临的挑战2018-08-29 22230
-
“建物致知”的合成生物学!引领“第三次生物科技革命”的合成生物学2018-08-10 18753
-
DNA纳米结构的功能化及其生物学应用2018-02-11 928
-
基于biosql构建了分子生物学二次数据库2017-12-12 1490
-
基于XML的生物学远程诊断系统2009-09-02 838
-
INA102心电图检测、记录生物学信号电路图2009-06-25 1479
-
食品卫生微生物学检验 菌落总数测定 GB/T 4789.2-2008-12-25 1102
全部0条评论

快来发表一下你的评论吧 !
