

 用磷酸揭示氮化硅对二氧化硅的选择性蚀刻机理
用磷酸揭示氮化硅对二氧化硅的选择性蚀刻机理
今日头条
描述
关键词:氮化硅,二氧化硅,磷酸,选择性蚀刻,密度泛函理论,焦磷酸
介绍
信息技术给我们的现代社会带来了巨大的转变。为了提高信息技术器件的存储密度,我们华林科纳使用浅沟槽隔离技术将半导体制造成无漏电流的极端规模集成。在这个过程中,固相氮化硅(Si3N4)层在部分二氧化硅(SiO2)沉积中起到掩模的作用。通过这种沉积,形成了由数百个交替堆叠的Si3N4和二氧化硅原子层组成的垂直堆叠结构.Si3N4掩模必须在程序结束时去除,通常通过热化学蚀刻。因此,在二氧化硅上选择性和完全蚀刻Si3N4是STI技术中制造高性能半导体器件的关键步骤。
典型地,选择性被定义为Si3N4和二氧化硅的蚀刻速率之间的比率。然而,由于传统蚀刻剂对两种硅材料的优先化学亲和力的边际差异,选择性蚀刻相当具有挑战性。此外,这两种材料在标准温度和压力下都是化学惰性的。在当代方法中,蚀刻在两种不同的条件下进行:干蚀刻和湿蚀刻。干法蚀刻通过离子轰击物理去除材料。众所周知,由于底切的各向异性和可忽略的趋势,产生高分辨率蚀刻。然而,由于不希望的低选择性,这是不利的,因为离子随机攻击表面,甚至损坏基底。另一方面,湿法蚀刻显示出比干法更高的选择性,对衬底的损坏程度更小,更适合大规模生产。因此,在各种商业制造技术中通常采用湿法蚀刻。
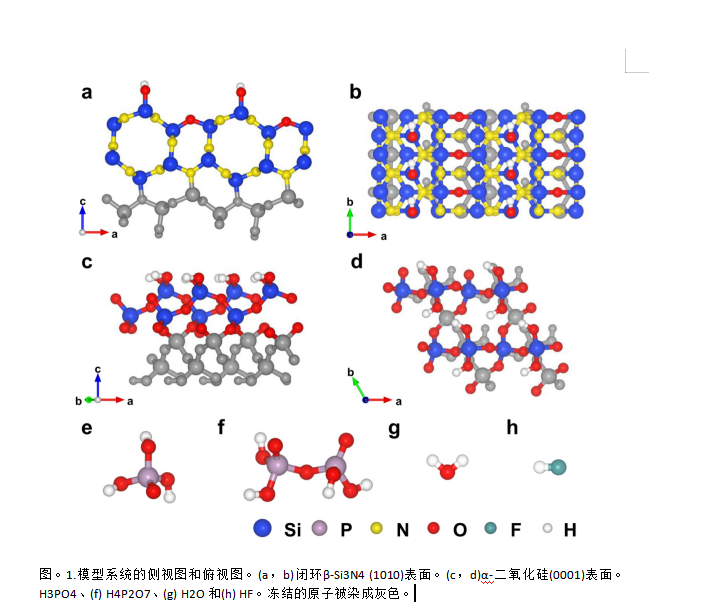
讨论
随着温度的升高,等式(1)中的平衡明显向右移动,这意味着溶液中大量生成。拉曼和核磁共振实验进一步观察到缩合焦磷酸的普遍存在。
在很大程度上,两种磷酸和水的相互作用是如何对二氧化硅层上的Si3N4进行选择性蚀刻的仍不清楚。在本研究中,我们阐明了H4P2O7和H3PO4分别对Si3N4和SiO2进行高选择性化学刻蚀的原子机制。密度泛函理论(DFT)计算被用于热力学识别一系列选择性蚀刻工艺的可行反应路径。在连续反应中,我们确定了一个速率决定步骤:Si–N和Si–O的化学键分别在Si3N4和SiO2表面解离。这些行为使用更多的奥费拉尔-詹克斯(MOFJ)图进一步证明。我们还研究了氟化氢作为蚀刻剂,以比较它与二氧化硅和氮化硅的相对反应性,因为二氧化硅与氟化氢的反应性比与磷酸的反应性更强。[20]此外,通过溶液中的质子传输,我们解耦了水分子对蚀刻过程的催化作。
计算方法 略
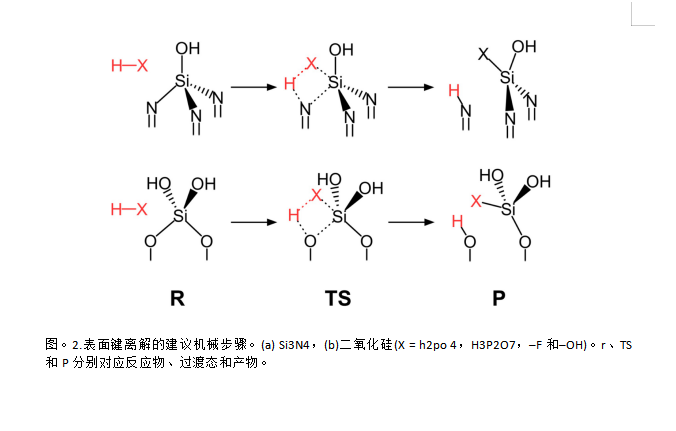
Si3N4的顺序蚀刻
顺序蚀刻的剩余部分依赖于热力学论证,假设它们的动力学根据贝尔-埃文斯波兰伊(BEP)原理与热力学相关。表1总结了预期的顺序反应及其能量,图2给出了相应的示意图。6.在通过H4P2O7 (R1)断开硅-氮键之后,应该断开另外两个硅-氮键以完成一个Si3N4蚀刻循环.然而,与硅原子结合的H4P2O7分子阻止另一蚀刻剂分子接近,因为其大分子结构引起空间位阻因此,随后的反应被延迟。H2O分子(R2*)取代连接的H4P2O7被计算为热力学上不可行(+0.43电子伏),而焦磷酸部分消除磷酸基团(R2)的水解是有利的(-0.31电子伏)。因此,H3PO4水解排放更可行。将键离解和水解过程放在一起,连续的反应分为两个步骤(R3和R4)。两个反应都是放热反应,反应能分别为-1.18和-1.58 eV。这意味着根据BEP原理,活化能低于第一步。虽然我们没有明确计算R3和R4的活化能,但反应热的增加趋势支持了我们的假设,即使用第一个硅氮键断裂作为限速步骤。总的来说,整个连续反应步骤充分描述了Si3N4全蚀刻循环的热力学可行性。
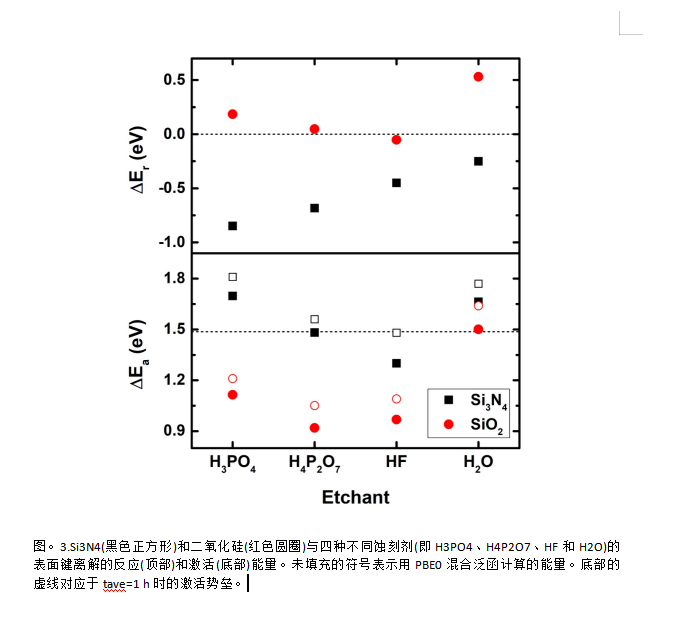
结论
本文研究了浓磷酸水溶液对氮化硅和二氧化硅的腐蚀机理。我们的结果清楚地表明,腐蚀性蚀刻剂对Si3N4具有很强的选择性,这对于各种半导体器件的表面工程是必不可少的。我们揭示了基于密度泛函能量学的热力学和动力学模型的基本机制。重要的是,H4P2O7被认为是最具反应性的物种,在典型的蚀刻条件下,其形成在高浓度和温度下是热力学上有利的。由于更大的分子结构,H4P2O7可以更容易地向Si3N4和二氧化硅表面提供质子,从而显示出比H3PO4更高的反应性.这也与我们计算的离解过渡态的MOFJ图非常一致。由于对Si3N4的相对缓慢的动力学,不能确保水的选择性蚀刻.然而,水通过结构质子扩散机制降低了活化势垒,从而在蚀刻过程中发挥了重要作用。此外,水水解结合到表面的焦磷酸,以促进整个蚀刻循环的完成。
审核编辑:鄢孟繁
-
《氧化铝、碳化硅、氮化硅,谁才是工业陶瓷老大?》2026-04-29 897
-
晶圆背面二氧化硅边缘腐蚀的原因2025-07-09 1435
-
芯片制造中的二氧化硅介绍2025-04-10 6502
-
氮化硅薄膜制备方法及用途2024-11-24 3883
-
镀膜使用二氧化硅的作用2024-09-27 3098
-
磷酸的腐蚀特性及缓蚀剂 氮化硅湿法蚀刻中热磷酸的蚀刻率2022-08-30 7841
-
在超临界二氧化碳中蚀刻氧化硅薄膜2022-05-23 2245
-
二氧化硅蚀刻标准操作程序研究报告2022-03-10 2475
-
碳化硅和二氧化硅之间稳定性的刻蚀选择性2022-02-15 4526
-
用于CVD金刚石沉积的氮化硅表面预处理报告2022-01-21 1549
-
二氧化硅层在芯片中有何作用2021-12-21 13821
-
石灰石二氧化硅化验仪器设备系列2021-03-11 2146
-
PECVD工艺参数对二氧化硅薄膜致密性的影响2020-09-29 13059
全部0条评论

快来发表一下你的评论吧 !
