

深度解析机器学习模型加速传统DFT计算
描述
Pd40Ni10Cu30P20非晶合金具有非常出色的电催化活性并且在实验中表现出很优秀的耐用周期,这令它成为如今非常重要的析氢反应催化剂之一。然而,非晶合金建模困难、合金晶胞太大而引起的巨额DFT计算成本使得人们对其表现出如此优秀的催化性能的物理机理尚不清晰。
深圳大学张希老师课题组建立了Smooth Overlap of Atomic Positions-Machine Learning (SOAP-ML)模型加速传统DFT计算并探究局部化学环境对于Pd40Ni10Cu30P20催化性能的影响。
利用该模型能够采集大量的活性位点(40000个活性位点)并精确地预测出具有最佳催化性能的非晶合金局部化学环境(该模型的能量预测值与DFT计算值的均方差为0.018)。
该论文“Exploring the physical origin of the electrocatalytic performanceof an amorphous alloy catalyst via machine learning accelerated DFT study”发表在《Nanoscale》期刊上。
SOAP-ML模型的算法框架分为三部分:
(i) 在非晶合金上、下表面随机生成540个活性位点样本,DFT计算得到精确的吉布斯自由能 并构建原始数据集;
(ii) 利用SOAP算符进行特征提取,构建吉布斯自由能的机器学习势函数并分析局域化学环境和吉布斯自由能的关系;
(iii) 在非晶合金的上、下表面密集采样各20000个活性位点,通过机器学习函数预测位点的吉布斯自由能,得到具有最佳电催化性能的局域原子配比。
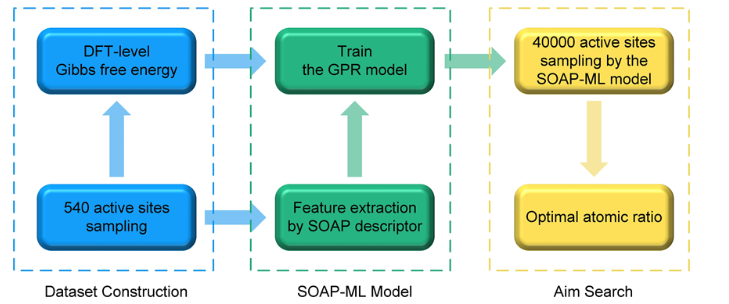
图1 SOAP-ML算法模型流程图
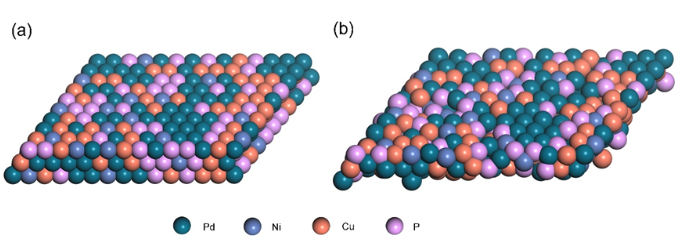
图2 (a) Pd40Ni10Cu30P20非晶合金优化前的结构;(b) DFT优化后的Pd40Ni10Cu30P20结构
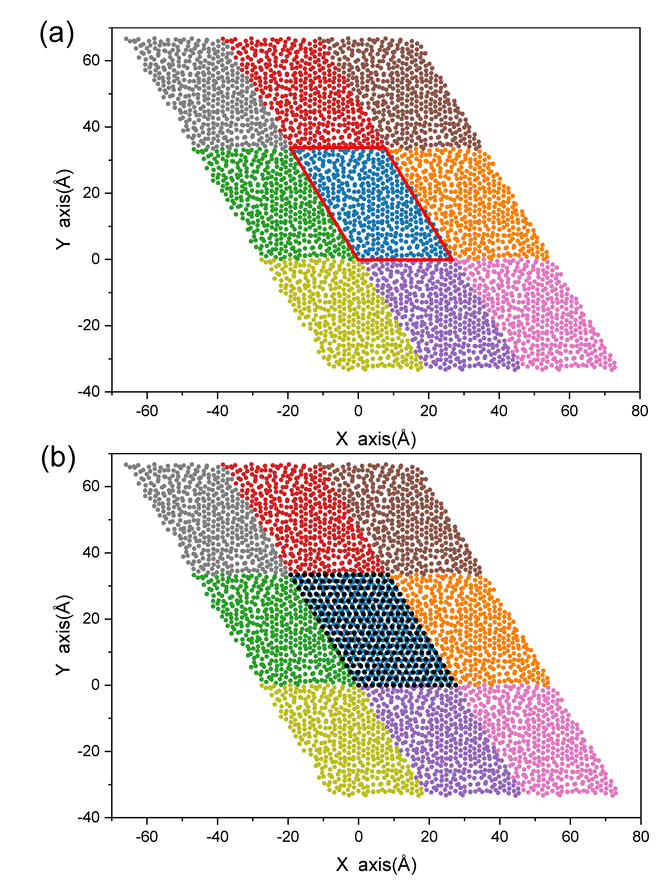
图3 (a) 3×3Pd40Ni10Cu30P20非晶合金的俯视图,其中红色框表示合金的单胞,用不同颜色标记每个单胞;(b) 活性位点的采样分布俯视图
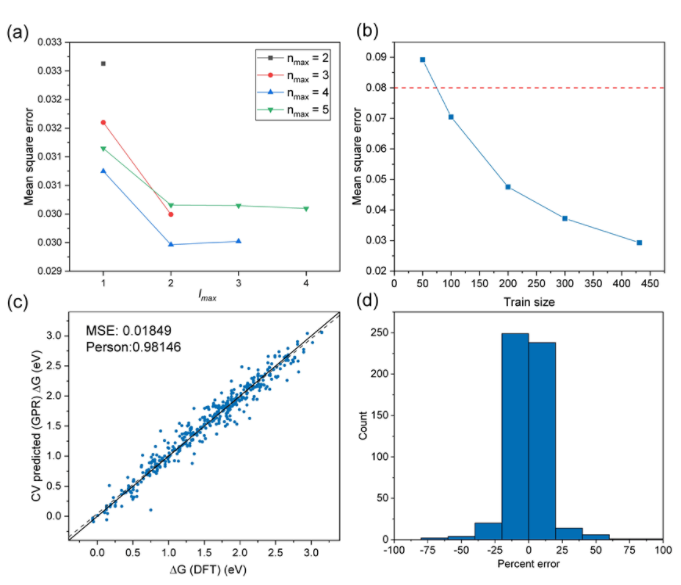
图4 (a) 不同lmax和nmax下对应的均方差值;(b) 训练集规模与均方差的关系;(c) DFT计算与用GPR模型预测得到的吉布斯自由能的机器学习交叉验证;(d) 模型预测的相对误差百分比
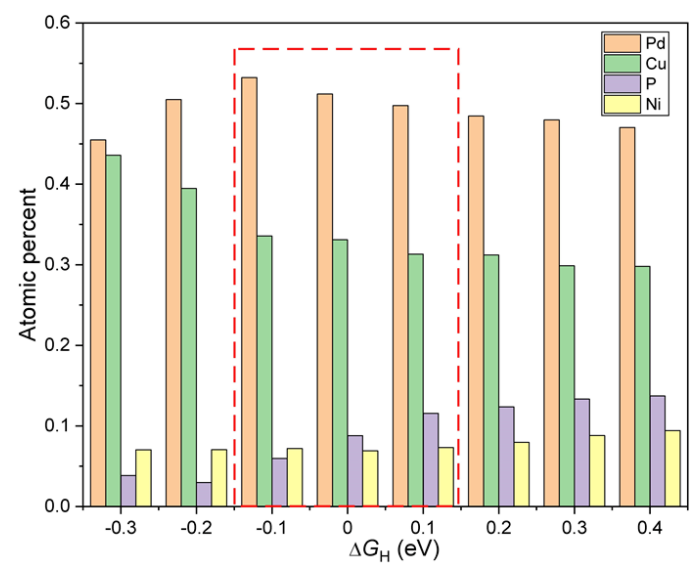
图5 不同吉布斯自由能下局域化学环境的原子百分比
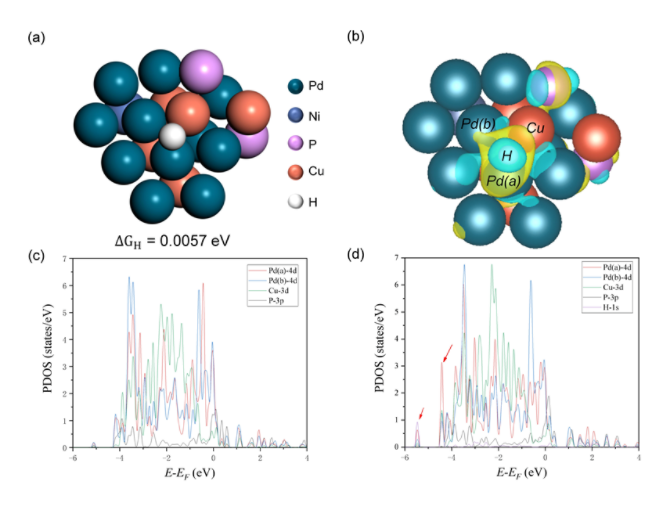
图6 (a) 的活性位点的原子结构模型;(b) 氢吸附前后的差分电荷密度,黄色和蓝色分别表示电荷累积和电荷损失;(c) 活性位点氢吸附前的PDOS图;(d) 活性位点氢吸附后的PDOS图
基于SOAP-ML模型,该团队筛选出吉布斯自由能位于范围内的3236个活性位点并进行分析,得到了局域化学环境的最佳配比Pd : Cu : P : Ni = 0.51 : 0.33 : 0.09 : 0.07。分析出Pd和Cu原子对合金局域化学环境的催化性能有促进作用,而P原子对活性位点的催化性能有抑制作用。
此外,该团队证实了Pd40Ni10Cu30P20非晶合金的长耐用周期来源于脱镍。
该团队进一步使用DFT计算了活性位点氢吸附前后的差分电荷密度与PDOS图,揭示了Pd原子与Cu原子对析氢反应起促进作用的机理。
该团队提出的SOAP-ML模型能够精准预测具有最佳催化性能的活性位点原子配比,其机器学习模型预测的结果达到了与DFT结果相比非常高的精度(其均方误差为0.018)。
这些结果为今后高性能非晶合金催化材料的设计提供了系统性的参考,并对非晶合金的局域化学环境与吉布斯自由能的关系提供了比较深刻的见解。
深圳大学高思妍与甄惠杰为论文的共同第一作者,本论文的通讯作者为深圳大学张希老师,深圳大学机电学院马将教授为合作者,深圳大学为第一完成单位。该项研究受到国家自然科学基金委以及深圳市海外高层次人才创新创业计划的资助,荔园优青第二期资助。
-
FPGA加速深度学习模型的案例2024-10-25 2282
-
AI大模型与传统机器学习的区别2024-10-23 4319
-
深度学习与传统机器学习的对比2024-07-01 4252
-
机器学习和深度学习的区别2023-08-28 2389
-
如何使用FPGA加速深度学习计算?2023-03-09 3755
-
大模型为什么是深度学习的未来?2023-02-16 3158
-
TDA4对深度学习的重要性2022-11-03 1512
-
什么是机器学习? 机器学习基础入门2022-06-21 3055
-
深度学习模型是如何创建的?2021-10-27 2392
-
为什么说FPGA是机器深度学习的未来?2019-10-10 3769
-
【瑞芯微RK1808计算棒试用申请】基于机器学习的视觉机械臂研究与设计2019-09-23 1577
-
深度学习模型压缩与加速综述2019-06-08 6347
-
Xilinx FPGA如何通过深度学习图像分类加速机器学习2018-11-28 4641
-
图像分类的方法之深度学习与传统机器学习2017-09-28 1806
全部0条评论

快来发表一下你的评论吧 !
