

QM/MM几何构型优化计算的python脚本
描述
QM/MM结构优化
QM/MM几何构型优化计算的python脚本如下:
import glob, math, os.path
from pBabel import AmberCrdFile_ToCoordinates3,
AmberTopologyFile_ToSystem ,
SystemGeometryTrajectory ,
AmberCrdFile_FromSystem ,
PDBFile_FromSystem ,
XYZFile_FromSystem
from pCore import Clone, logFile, Selection
from pMolecule import NBModelORCA, QCModelBDF, System
from pMoleculeScripts import ConjugateGradientMinimize_SystemGeometry,
FIREMinimize_SystemGeometry ,
LBFGSMinimize_SystemGeometry ,
SteepestDescentMinimize_SystemGeometry
# 定义结构优化接口
def opt_ConjugateGradientMinimize(molecule, selection):
molecule.DefineFixedAtoms(selection) #固定原子
#定义优化方法
ConjugateGradientMinimize_SystemGeometry(
molecule,
maximumIterations = 40, # 最大优化步数
rmsGradientTolerance = 0.1, #优化收敛控制
trajectories = [(trajectory, 1)]
) # 定义轨迹保存频率
# 定义能量计算模式
nbModel = NBModelORCA()
qcModel = QCModelBDF("GB3LYP:6-31g")
# 读取体系坐标和拓扑信息
molecule = AmberTopologyFile_ToSystem ("GallicAcid.prmtop")
molecule.coordinates3 = AmberCrdFile_ToCoordinates3("GallicAcid.crd")
# 关闭体系对称性
molecule.DefineSymmetry(crystalClass = None) # QM/MM方法不支持使用周期性边界条件
#. Define Atoms List
natoms = len(molecule.atoms) # 系统中总原子数
qm_list = range(72, 90) # QM 区原子
activate_list = range(126, 144) + range (144, 162) # MM区活性原子(优化中可以移动)
#定义MM区原子
mm_list = range (natoms)
for i in qm_list:
mm_list.remove(i) # MM 删除QM原子
mm_inactivate_list = mm_list[:]
for i in activate_list :
mm_inactivate_list.remove(i)
# 输入QM原子
qmmmtest_qc = Selection.FromIterable(qm_list)
# 定义各选择区
selection_qm_mm_inactivate = Selection.FromIterable(qm_list + mm_inactivate_list)
selection_mm = Selection.FromIterable(mm_list)
selection_mm_inactivate = Selection.FromIterable(mm_inactivate_list)
# . Define the energy model.
molecule.DefineQCModel(qcModel, qcSelection = qmmmtest_qc)
molecule.DefineNBModel(nbModel)
molecule.Summary()
#计算优化开始时总能量
eStart = molecule.Energy()
#定义输出文件目录名
outlabel = 'opt_watbox_bdf'
if os.path.exists(outlabel):
pass
else:
os.mkdir (outlabel)
outlabel = outlabel + '/' + outlabel
# 定义输出轨迹
trajectory = SystemGeometryTrajectory (outlabel + ".trj" , molecule, mode = "w")
# 开始第一阶段优化
# 定义优化两步
iterations = 2
# 顺次固定QM区和MM区进行优化
for i in range(iterations):
opt_ConjugateGradientMinimize(molecule, selection_qm_mm_inactivate) #固定QM区优化
opt_ConjugateGradientMinimize(molecule, selection_mm) #固定MM区优化
# 开始第二阶段优化
# QM区和MM区同时优化
opt_ConjugateGradientMinimize(molecule, selection_mm_inactivate)
#输出优化后总能量
eStop = molecule.Energy()
#保存优化坐标,可以为xyz/crd/pdb等。
XYZFile_FromSystem(outlabel + ".xyz", molecule)
AmberCrdFile_FromSystem(outlabel + ".crd" , molecule)
PDBFile_FromSystem(outlabel + ".pdb" , molecule)
输出体系收敛信息如下(此处仅展示前20步优化收敛结果):
----------------------------------------------------------------------------------------------------------------
Iteration Function RMS Gradient Max. |Grad.| RMS Disp. Max. |Disp.|
----------------------------------------------------------------------------------------------------------------
0 I -1696839.69778731 2.46510318 9.94250232 0.00785674 0.03168860
2 L1s -1696839.82030342 1.38615730 5.83254788 0.00043873 0.00126431
4 L1s -1696839.90971371 1.41241184 5.29242524 0.00067556 0.00172485
6 L0s -1696840.01109863 1.41344485 4.70119338 0.00090773 0.00265969
8 L1s -1696840.09635696 1.44964059 5.72496661 0.00108731 0.00328490
10 L1s -1696840.17289698 1.28607709 4.73666387 0.00108469 0.00354577
12 L1s -1696840.23841524 1.03217304 3.00441004 0.00081945 0.00267931
14 L1s -1696840.30741088 1.40349698 5.22220965 0.00162080 0.00519590
16 L1s -1696840.43546466 1.32604042 4.51175225 0.00158796 0.00455431
18 L0s -1696840.52547251 1.27123125 4.20616166 0.00158796 0.00428040
20 L0s -1696840.60265453 1.08553355 3.12355616 0.00158796 0.00470223
----------------------------------------------------------------------------------------------------------------
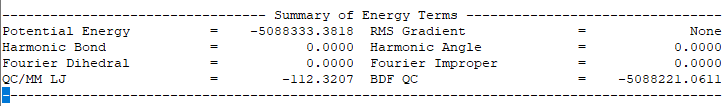
输出体系总能量信息如下:

注:QM/MM几何构型优化一般不容易收敛,在实际操作中需要的技巧较多。常见的有,固定MM区,优化QM区;然后固定QM区优化MM区。如此往复循环几次后,再同时优化QM区和MM区。优化是否收敛,和QM区的选择及QM/MM边界是否有带电较多的原子等关系很大。为了加速优化,可以在计算时固定MM区,仅选择离QM区较近的合适区域,作为活性区域,在优化中坐标可以变化。
QM/MM激发态计算
基于上一步的QM/MM几何构型优化,继而即可将MM区活性原子添加到QM区进行QM/MM-TDDFT计算,完整的代码如下:import glob, math, os.path
from pBabel import AmberCrdFile_ToCoordinates3,
AmberTopologyFile_ToSystem ,
SystemGeometryTrajectory ,
AmberCrdFile_FromSystem ,
PDBFile_FromSystem ,
XYZFile_FromSystem
from pCore import Clone, logFile, Selection
from pMolecule import NBModelORCA, QCModelBDF, System
from pMoleculeScripts import ConjugateGradientMinimize_SystemGeometry,
FIREMinimize_SystemGeometry ,
LBFGSMinimize_SystemGeometry ,
SteepestDescentMinimize_SystemGeometry
# 定义结构优化接口
def opt_ConjugateGradientMinimize(molecule, selection):
molecule.DefineFixedAtoms(selection) #固定原子
#定义优化方法
ConjugateGradientMinimize_SystemGeometry(
molecule,
maximumIterations = 40, # 最大优化步数
rmsGradientTolerance = 0.1, #优化收敛控制
trajectories = [(trajectory, 1)]
) # 定义轨迹保存频率
# 定义能量计算模式
nbModel = NBModelORCA()
qcModel = QCModelBDF("GB3LYP:6-31g")
# 读取体系坐标和拓扑信息
molecule = AmberTopologyFile_ToSystem ("GallicAcid.prmtop")
molecule.coordinates3 = AmberCrdFile_ToCoordinates3("GallicAcid.crd")
# 关闭体系对称性
molecule.DefineSymmetry(crystalClass = None) # QM/MM方法不支持使用周期性边界条件
#. Define Atoms List
natoms = len(molecule.atoms) # 系统中总原子数
qm_list = range(72, 90) # QM 区原子
activate_list = range(126, 144) + range (144, 162) # MM区活性原子(优化中可以移动)
#定义MM区原子
mm_list = range (natoms)
for i in qm_list:
mm_list.remove(i) # MM 删除QM原子
mm_inactivate_list = mm_list[:]
for i in activate_list :
mm_inactivate_list.remove(i)
# 输入QM原子
qmmmtest_qc = Selection.FromIterable(qm_list)
# 定义各选择区
selection_qm_mm_inactivate = Selection.FromIterable(qm_list + mm_inactivate_list)
selection_mm = Selection.FromIterable(mm_list)
selection_mm_inactivate = Selection.FromIterable(mm_inactivate_list)
# . Define the energy model.
molecule.DefineQCModel(qcModel, qcSelection = qmmmtest_qc)
molecule.DefineNBModel(nbModel)
molecule.Summary()
#计算优化开始时总能量
eStart = molecule.Energy()
#定义输出文件目录名
outlabel = 'opt_watbox_bdf'
if os.path.exists(outlabel):
pass
else:
os.mkdir (outlabel)
outlabel = outlabel + '/' + outlabel
# 定义输出轨迹
trajectory = SystemGeometryTrajectory (outlabel + ".trj" , molecule, mode = "w")
# 开始第一阶段优化
# 定义优化两步
iterations = 2
# 顺次固定QM区和MM区进行优化
for i in range(iterations):
opt_ConjugateGradientMinimize(molecule, selection_qm_mm_inactivate) #固定QM区优化
opt_ConjugateGradientMinimize(molecule, selection_mm) #固定MM区优化
# 开始第二阶段优化
# QM区和MM区同时优化
opt_ConjugateGradientMinimize(molecule, selection_mm_inactivate)
#输出优化后总能量
eStop = molecule.Energy()
#保存优化坐标,可以为xyz/crd/pdb等。
XYZFile_FromSystem(outlabel + ".xyz", molecule)
AmberCrdFile_FromSystem(outlabel + ".crd" , molecule)
PDBFile_FromSystem(outlabel + ".pdb" , molecule)
# TDDFT计算
qcModel = QCModelBDF_template ( )
qcModel.UseTemplate (template = 'head_bdf_nosymm.inp' )
tdtest = Selection.FromIterable ( qm_list + activate_list )
# . Define the energy model.
molecule.DefineQCModel ( qcModel, qcSelection = tdtest )
molecule.DefineNBModel ( nbModel )
molecule.Summary ( )
# . Calculate
energy = molecule.Energy ( )
输出体系总能量信息如下:
No. 1 w= 4.7116 eV -1937.8276358207 a.u. f= 0.0217 D= 0.0000 Ova= 0.6704
CV(0): A( 129 )-> A( 135 ) c_i: 0.7254 Per: 52.6% IPA: 7.721 eV Oai: 0.6606
CV(0): A( 129 )-> A( 138 ) c_i: 0.2292 Per: 5.3% IPA: 9.104 eV Oai: 0.8139
CV(0): A( 132 )-> A( 135 ) c_i: 0.4722 Per: 22.3% IPA: 7.562 eV Oai: 0.6924
CV(0): A( 132 )-> A( 138 ) c_i: -0.4062 Per: 16.5% IPA: 8.946 eV Oai: 0.6542
随后还打印了跃迁偶极矩
*** Ground to excited state Transition electric dipole moments (Au) ***
State X Y Z Osc.
1 0.0959 0.1531 0.3937 0.0217 0.0217
2 0.0632 -0.1286 0.3984 0.0207 0.0207
3 -0.0797 -0.2409 0.4272 0.0287 0.0287
4 0.0384 -0.0172 -0.0189 0.0003 0.0003
5 1.1981 0.8618 -0.1305 0.2751 0.2751
吸收光谱分析
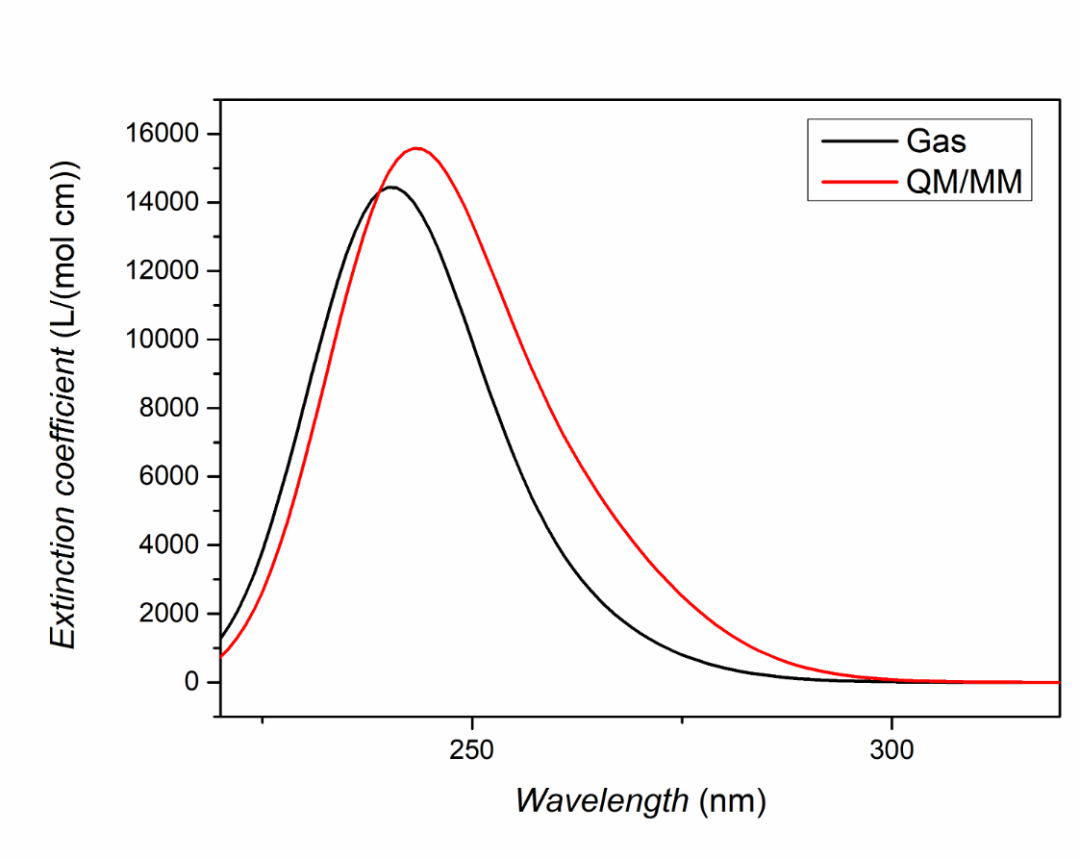
对于激发态我们往往需要得到理论预测的吸收谱峰形,也就是将每个激发态的吸收按一定的半峰宽进行高斯展宽。在TDDFT计算正常结束后,我们需要进入终端用命令调用BDF安装路径下的plotspec.py脚本执行计算。若用户使用鸿之微云算力资源,进入命令端方式请查阅鸿之微云指南,此文不做赘述。
进入终端后,在目录下运行$BDFHOME/sbin/plotspec.py bdf.out,会产生两个文件,分别为bdf.stick.csv和bdf.spec.csv,前者包含所有激发态的吸收波长和摩尔消光系数,可以用来作棒状图,后者包含高斯展宽后的吸收谱(默认的展宽FWHM为0.5 eV),分别对比真空环境以及溶剂化效应下高斯展宽后的吸收谱情况,并用Excel、Origin等作图软件作图如下:
-
如何在 IIS 中执行 Python 脚本2010-02-23 1582
-
python编写脚本方法2017-11-17 5347
-
如何使html网页与python脚本进行通信2019-11-04 8545
-
基于Python脚本的R语言的函数2020-10-12 2891
-
基于Python的实时嵌入式软件测试脚本总结2021-07-30 1430
-
【Python】如何将Python脚本打包成exe可执行文件2022-08-18 19991
-
通过Python脚本实现WIFI密码的暴力破解2022-09-19 8173
-
揭示尖晶石催化剂几何构型影响HER活性的机理2022-11-03 5615
-
10个杀手级的Python自动化脚本2022-11-28 1176
-
13个用于日常编程的高级Python脚本2022-12-09 1046
-
分享Perl和Python脚本轻量实用的调试工具2023-01-21 2904
-
将Python脚本集成到GUI工具包2023-02-15 2104
-
Python怎么玩转JS脚本2023-02-23 2446
-
如何在Linux命令行中运行Python脚本2023-05-12 2987
-
通过Python脚本实现WIFI密码的自动猜解2024-01-25 5140
全部0条评论

快来发表一下你的评论吧 !
