

两种络合物电子结构和光化学性质
描述
01 引言
单态氧是一种处于激发态的活性氧,具有独特的电子结构和较强的氧化性,并且氧化后不产生有毒有害的副产物,属于绿色、环境友好型氧化剂,在医学、生物学和大气化学等领域均发挥重要作用,使得关于单态氧的研究成为经久不衰的课题。但自然界中的氧分子是以三重态形式存在的,单重态氧则需要通过实验手段才能获得,例如内过氧化物的还原、光敏化方法和超声波方法。然而,这些方法在实际应用中都面临着诸多困难。因此,人们试图寻找可在常温便捷的实验条件下产生单态氧的方法。因为在比较温和的条件下生成单态氧的反应,可减少外加条件(如光、加热和超声等)在应用中的干扰、减少操作过程的复杂度并节约相应的能源。
李明德课题组发现在室温无光照的条件下仅通过蒽醌和叔丁醇钾的络合物(AQ@[K(O-tBu)]4)即可催化三态氧生成单态氧,而且该络合物经蓝光或绿光照射后的产物(AQ@[K(O-tBu)]4*)同样可以催化三态氧生成单态氧。这一发现是令人兴奋的,因为通常蒽醌类化合物是需要光照的条件才可产生单态氧或其他活性氧物种的,这一发现则为单态氧的合成提供了新的思路和便捷的实验手段。此外,蒽醌摆脱了传统观念中光敏剂的角色,以新的方式参与生成单态氧的反应,因此对于该反应机制的系统研究是十分重要的。基于这个目的,我们展开了这项工作。我们采用DFT/TDDFT和NEVPT2方法系统地研究了AQ@[K(O-tBu)]4和AQ@[K(O-tBu)]4*两种络合物的电子结构和光化学性质;通过MSDFT方法探究了反应中生成单态氧的电子结构;利用DFT方法合理推测了络合物催化三态氧生成单态氧的反应机理。本文工作为在常温且无光照的实验条件下产生单态氧的反应提供了理论依据。
02 成果简介
本文通过鸿之微的MOMAP软件,采用DFT/TDDFT和NEVPT2方法系统地研究了AQ@[K(O-tBu)]4和AQ@[K(O-tBu)]4*两种络合物的电子结构和光化学性质;通过MSDFT方法探究了两种络合物催化3O2生成1O2的电子结构;使用DFT方法从热力学角度合理地推测了络合物生成1O2可能的反应机理,所得结论如下:
1)室温且无光照条件下,AQ和K(O-tBu)的络合物为稳定的三重态络合物AQ@[K(O-tBu)]4。在络合过程中,当配体K(O-tBu)数达到3时络合物已经表现为三重态基态稳定。
2)络合物AQ@[K(O-tBu)]4和AQ@[K(O-tBu)]4*的反应中心均为蒽醌阴离子自由基,络合物可表示为离子化合物形式[AQ·]- [K(O-tBu)]4+。[AQ·]-的形成原因是由于配体中钾原子的电子具有强离域性。二者电子结构的差异表现在O-tBu部分的电子结构不同,在AQ@[K(O-tBu)]4中为π型,在AQ@[K(O-tBu)]4*中表现为σ型。两种络合物具有相同UV-Vis吸收峰的原因是因为二者激发模式相同,均为[AQ]-中单占据电子的激发,长波处均为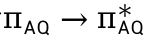 激发;短波处均为
激发;短波处均为 激发。
激发。
3)AQ@[K(O-tBu)]4络合物经光照转化为AQ@[K(O-tBu)]4*络合物的实质为:基态O-tBu自由基经光激发、振动弛豫后得到激发态的能量最小点结构(O-tBu)π*。(O-tBu)π*退回基态(O-tBu)π的辐射速率远慢于(O-tBu)π*经内转换形成(O-tBu)σ的无辐射速率,因此可以形成寿命较长的(O-tBu)σ,即络合物AQ@[K(O-tBu)]4经光照后快速形成激发态AQ@[K(O-tBu)]4,激发态AQ@[K(O-tBu)]4经振动弛豫、内转换形成稳定的三重态络合物AQ@[K(O-tBu)]4*。
4)反应物(3O2@[AQ·]-)与产物(1O2 (OS)@[AQ·]-)的透热耦合随着氧分子与[AQ·]-的距离减小而增大,距离为3.4Å时开始发生反应。反应物3O2@[AQ·]-与产物(1O2 (CS)@[AQ·]-)的透热耦合始终接近零。络合物AQ@[K(O-tBu)]4和AQ@[K(O-tBu)]4*催化3O2生成的1O2为开壳层,反应机理为3O2与@[AQ·]-的SOMO中电子交换。
经计算推测络合物AQ@[K(O-tBu)]4催化3O2生成的1O2的可能机理为:AQ@[K(O-tBu)]4与通入的3O2反应生成能量较低1[AQ@[K(O-tBu)]4@O2]络合物,此过程中结合氧一侧的两个K(O-tBu)有离去AQ@[K(O-tBu)]4的趋势,体系颜色褪去;继续向反应体系通入N2,将生成更稳定的络合物AQ@[K(O-tBu)]4@N2并释放1O2;停止通入N2时,由于N2不易溶于水而从体系中离去,有离去趋势的两个K(O-tBu)重新结合为稳定的基态三重态络合物AQ@[K(O-tBu)]4,体系也变为最初体系的颜色。
03 图文导读
3.1 AQ@[K(O-tBu)]4络合物
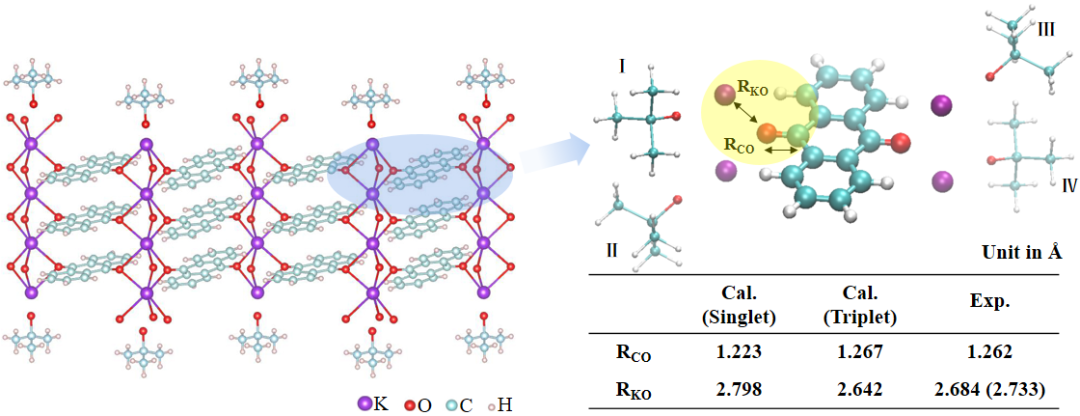
图1 左侧为AQ@[K(O-tBu)]4络合物晶体结构。右上方为AQ@[K(O-tBu)]4络合物的计算模型。4个K(O-tBu)分别标记为Ⅰ、Ⅱ、Ⅲ和Ⅳ。表中为已优化单、三重态AQ@[K(O-tBu)]4结构和实验晶体中C=O键长(RCO)及AQ的C=O键中O与K(O-tBu)中K的距离(RKO)。
3.2 AQ@[K(O-tBu)]4络合物中阴离子自由基([AQ·]-)及吸收光谱计算
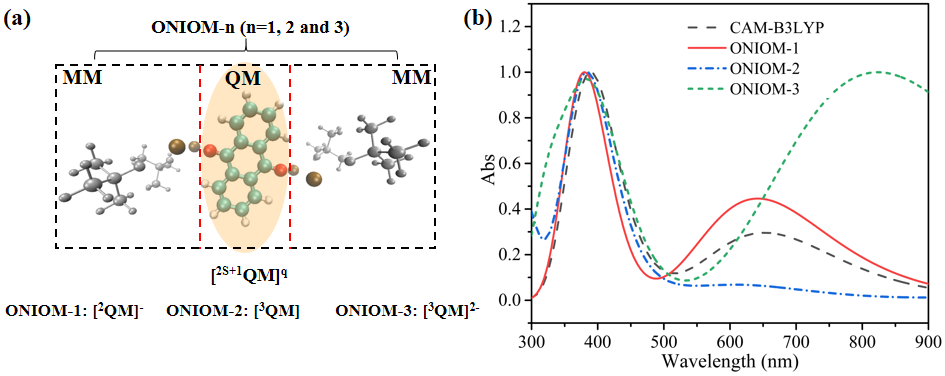
图2 (a) AQ@[K(O-tBu)]4络合物的ONIOM模型。橙色的AQ部分为QM区域,灰色部分为MM区域。“2S+1”为QM区域的自旋多重度。“q”为QM区域所带的电荷。(b) 不同ONIOM模型及CAM-B3LYP结果的UV-Vis。其中,ONIOM-1用红色实线表示,ONIOM-2使用蓝色虚线表示,ONIOM-3使用绿色虚线表示,CAM-B3LYP结果标记为黑色虚线。
3.3 借助MOMAP计算不同自由基结构之间的跃迁过程
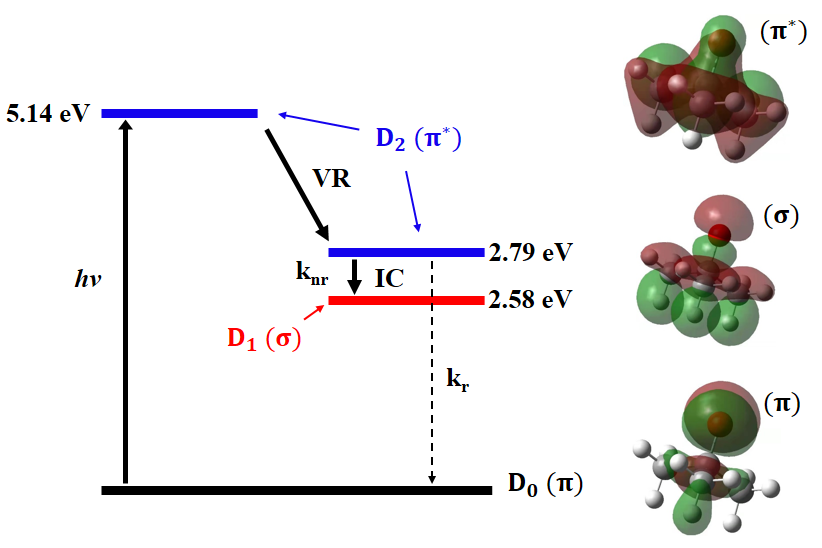
图3 O-tBu自由基的跃迁能级图。右侧为基态(D0,π)、第一激发态(D1,
σ)和第二激发态(D2,π*)的电子结构。knr为D2→D1的无辐射跃迁速率(2.02×1018 s-1),kr为D2→D0的辐射跃迁速率(2.58×107 s-1)。
3.4 络合物AQ@[K(O-tBu)]4和AQ@[K(O-tBu)]4*催化三态氧生成单态氧的反应机理
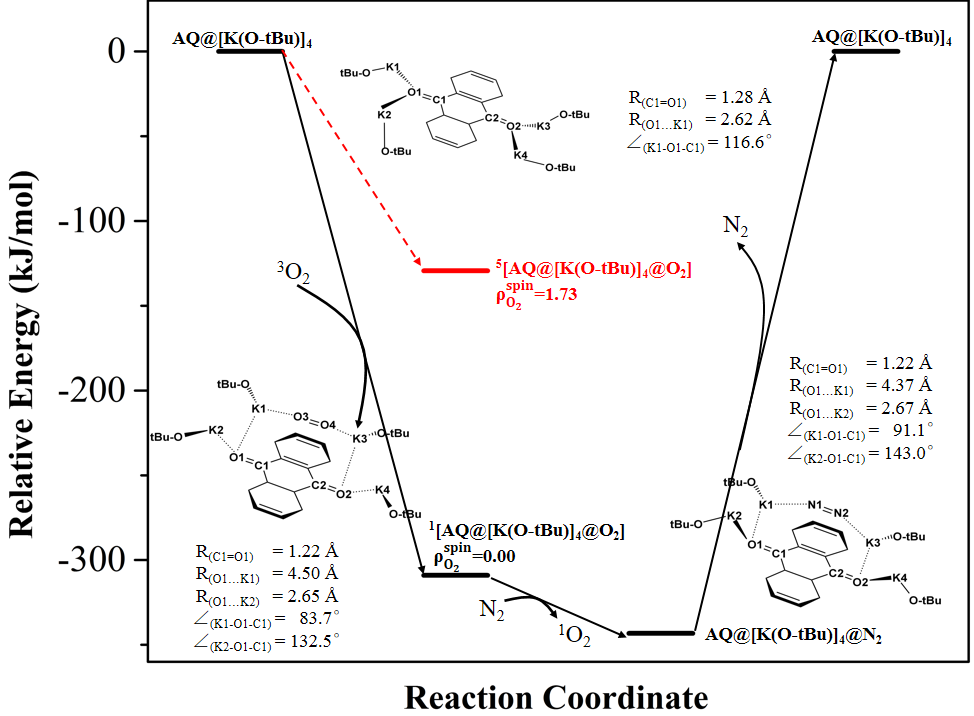
图4 络合物AQ@[K(O-tBu)]4催化3O2生成1O2的反应坐标图,络合物AQ@[K(O-tBu)]4、1[AQ@[K(O-tBu)]4@O2]和AQ@[K(O-tBu)]4@N2的结构。
04 小结
本论文将借助密度泛函理论(DFT)和高精度从头算方法(NEVPT2)系统地研究AQ@[K(O-tBu)]4和AQ@[K(O-tBu)]4*的电子结构及光化学性质,并系统阐述这两种络合物催化三态氧生成单态氧的反应机理。结果表明这两种络合物均为稳定的三重态络合物,但其中钾原子中的电子表现出很强的弥散性,使得络合物产生了很强的电荷分离,这导致其中蒽醌部分带有一个负电荷,以蒽醌阴离子自由基的形式存在([AQ·]-)。进一步的研究发现,AQ@[K(O-tBu)]4与AQ@[K(O-tBu)]4*在电子结构上的差异仅为叔丁基部分电子结构的不同,其中,AQ@[K(O-tBu)]4的叔丁基电子结构为π型,AQ@[K(O-tBu)]4*的叔丁基电子结构为σ型。这一发现很好的解释了两种络合物具有相同的紫外可见吸收光谱的本质(二者的激发模式均为[AQ·]-中电子的激发)。此外,我们还采用多态密度泛函理论(MSDFT)对络合物催化产生单态氧的反应过程进行了研究,结果表明该络合物所催化产生的单态氧均具有开壳层电子结构。本论文的工作揭示了AQ@[K(O-tBu)]4化合物催化产生单态氧微观机制,为蒽醌类化合物在室温无光照条件下生成单态氧的实验提供了理论支撑,拓宽了蒽醌及其衍生物生成单态氧的途径。
-
金属材料的化学性能2017-08-25 2752
-
电化学原理介绍和分析方法2017-10-16 11892
-
电化学传感器的发展怎么样?2020-03-25 3306
-
光化学烟雾物理模拟实验数据采集系统的研制2009-06-29 537
-
一种新型的振荡场光化学传感器2009-07-14 693
-
光化学、干膜、曝光及显影制程2010-01-11 7437
-
光化学、干膜、曝光及显影制程术语手册2010-02-21 4458
-
创新光化学电池结合太阳能与化学储存2016-02-19 1934
-
机器学习的新量子化学工具可加速量子化学计算2020-10-16 2697
-
PCB板显影液的性质和处理方法2022-11-24 3712
-
了解半导体结的化学性质2023-01-05 2418
-
用于快速检测百草枯农药残留的纸基光化学传感器2023-02-03 652
-
什么锂电池的电化学性质和反应机理?看了你就明白了2023-02-24 3324
-
光化学反应量热仪(PDC)如何工作?2023-03-20 1873
-
镓的化学性质与应用2025-01-06 5639
全部0条评论

快来发表一下你的评论吧 !
