

MOMAP对azulene的S1→S0的辐射速率和内转换速率的计算结构优化
描述
我们使用BDF进行azulene的S1激发态结构优化和频率计算。选中Simulator → BDF → BDF,界面中设置参数。在Basic Settings界面中的Calculation Type选择TDDFT-OPT+Freq,方法采用默认的GB3LYP泛函,基组在Basis中的All Electron类型中,选择6-31G(d,p)。SCF Settings和TDDFT Settings面板中将Use MPEC+COSX Acceleraton的默认勾选去掉。Basic Settings、SCF Settings、TDDFT Settings界面中的其它参数以及OPT Settings、Freq Settings等面板的参数使用推荐的默认值,不需要做修改。之后点击 Generate files 即可生成对应计算的输入文件。
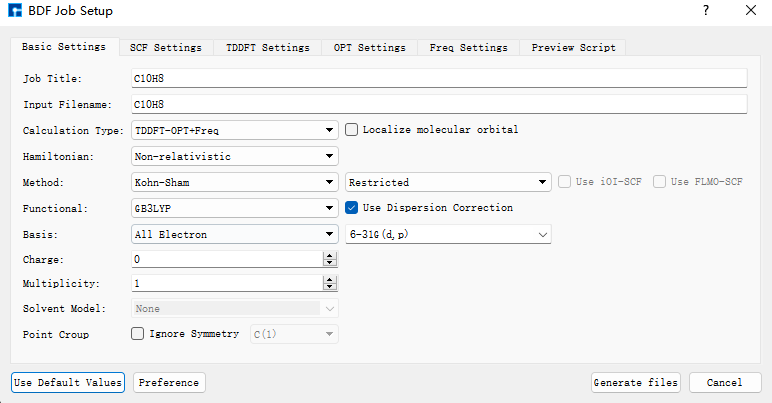
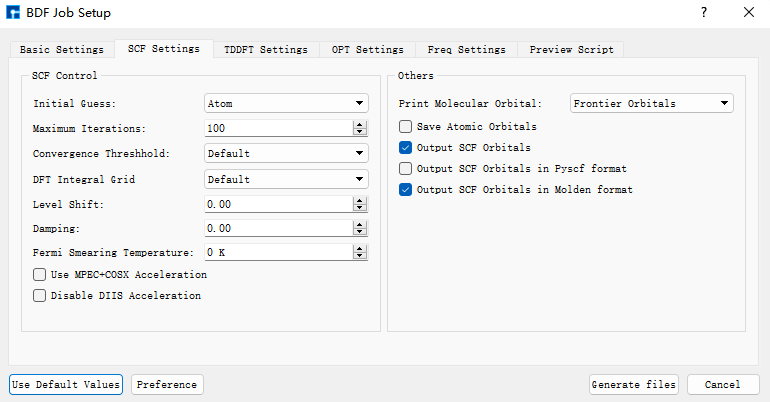
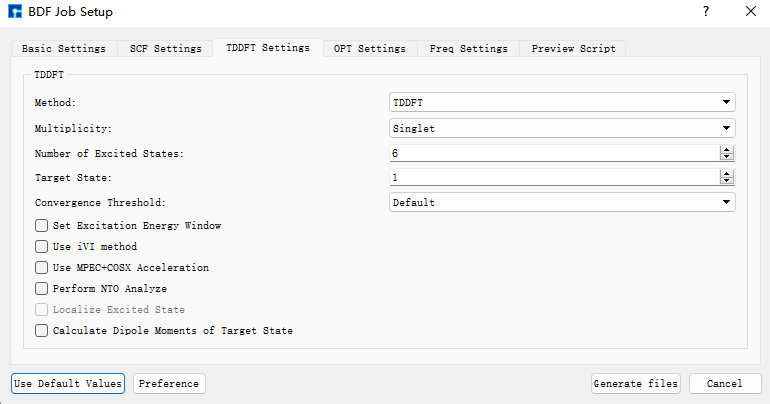
选中生成的bdf.inp文件,右击选择open with,打开该文件,如下所示:
$compass
Title
C10H8
Geometry
C 0.79273796 0.49102542 -0.00003307
C -0.70229649 0.61186591 0.00000000
C 1.30022932 1.80163337 -0.00006272
C -0.99262499 1.98726800 0.00007812
C 1.54415132 -0.67887418 0.00000000
C -1.63318173 -0.42094563 -0.00002837
C 0.21877157 2.69859813 0.00000000
C 1.10656346 -2.00562676 0.00005788
C -1.41619168 -1.80093044 -0.00004814
C -0.20258112 -2.49333483 0.00000000
H 2.35092512 2.06249889 -0.00009828
H -1.98777600 2.41348149 0.00017650
H 2.62424717 -0.53731745 0.00001117
H -2.67585843 -0.10561277 -0.00001521
H 0.30641472 3.77916915 0.00002386
H 1.88966566 -2.75951313 0.00017581
H -2.31053950 -2.41870505 -0.00009019
H -0.29054446 -3.57807510 0.00000000
End Geometry
Basis
6-31G(d,p)
Skeleton
Group
C(1)
$end
$bdfopt
Solver
1
MaxCycle
108
IOpt
3
Hess
final
$end
$xuanyuan
Direct
$end
$scf
RKS
Charge
0
SpinMulti
1
DFT
GB3LYP
D3
Molden
$end
$tddft
Imethod
1
Isf
0
Idiag
1
Iroot
6
Istore
1
$end
$resp
Geom
Method
2
Nfiles
1
Iroot
1
$end
选中bdf.inp文件,右击选择Run提交作业,任务结束后bdf.out,bdf.out.tmp,bdf.scf.molden三个结果文件会出现在Project中。 选择bdf.out,右击show view,在Optimization对话框中,显示结构已经达到收敛标准。
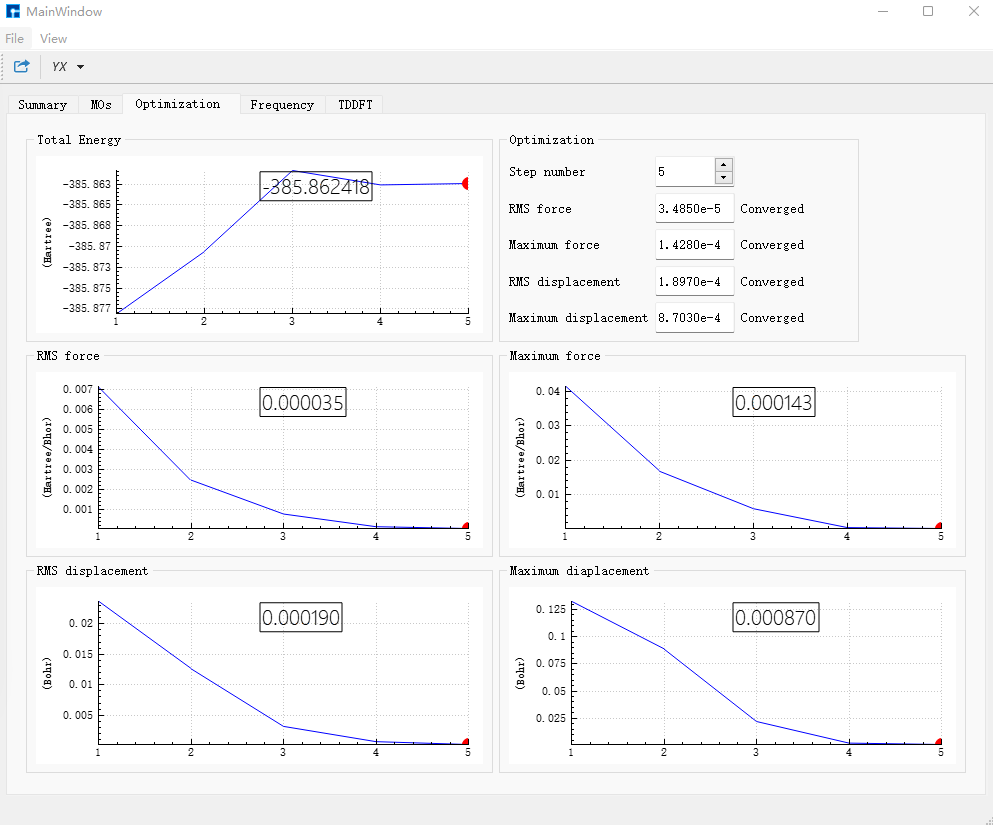
在Frequency对话框中,检查频率,若不存在虚频证明结构已经优化到极小点。
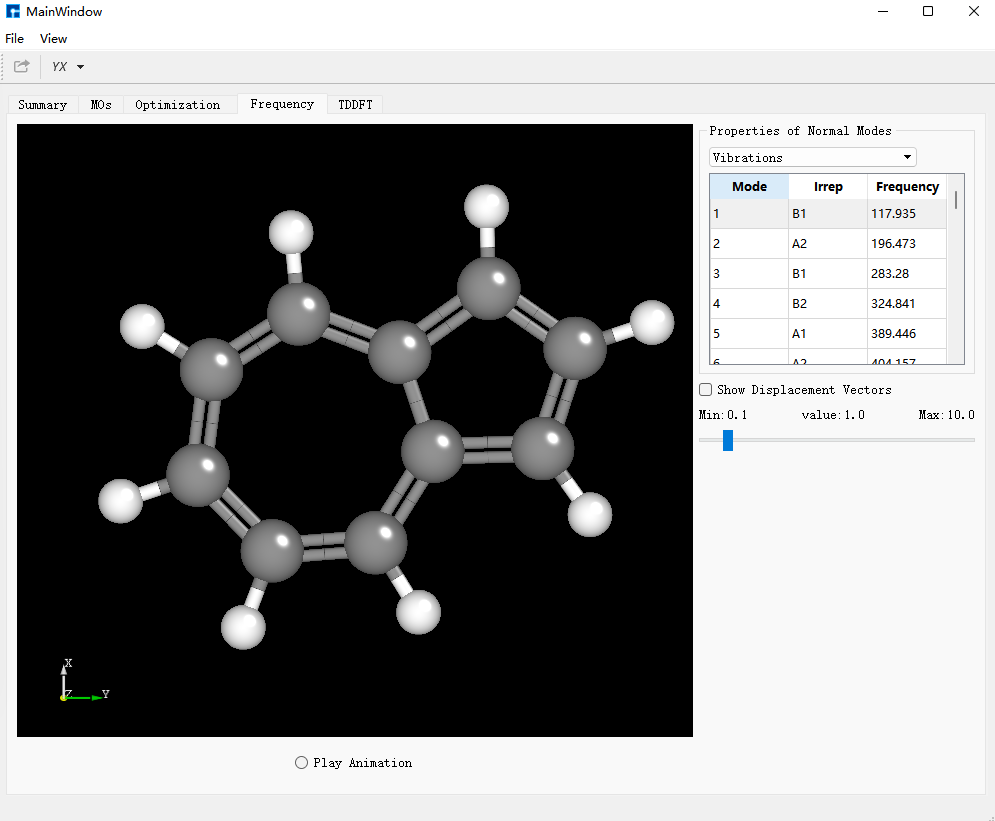
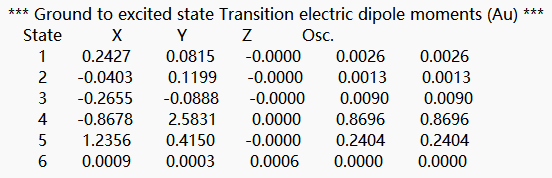
选择bdf.out.tmp,右击open with containing folder,打开bdf.out.tmp,在文件开始向下查找第一个tddft计算模块‘module tddft’。根据tddft模块的Ground to excited state Transition electric dipole moments (Au) 中的State 1的跃迁电偶极矩,得到momap需要的参数EDMA,计算方法为: 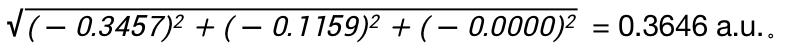 需要将单位a.u.转换为Debye,因此EDMA= 0.3646*2.5417=0.9267 Debye。
需要将单位a.u.转换为Debye,因此EDMA= 0.3646*2.5417=0.9267 Debye。
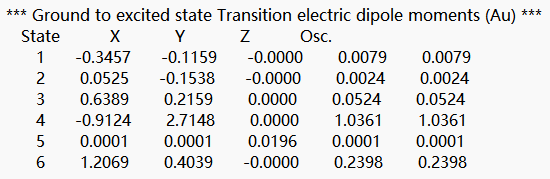
在文件末尾向上查找第一个tddft计算模块‘module tddft’。根据tddft模块的Ground to excited state Transition electric dipole moments (Au) 中的State 1的跃迁电偶极矩,得到momap需要的参数EDME,计算方法为: 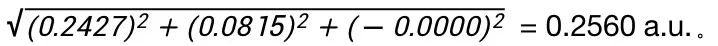 需要将单位a.u.转换为Debye,因此EDMA= 0.3646*2.5417=0.6507 Debye。
需要将单位a.u.转换为Debye,因此EDMA= 0.3646*2.5417=0.6507 Debye。
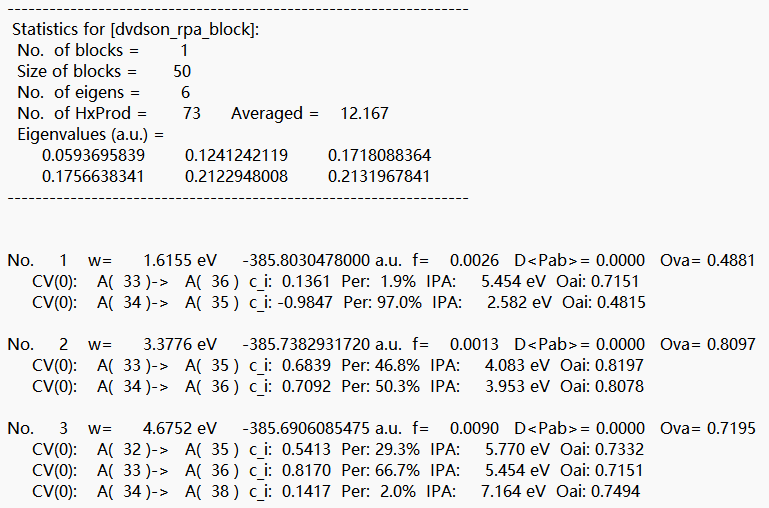
在该tddft模块的Statistics for [dvdson_rpa_block]:中读取No. 1态的能量为 -385.8030478000 a.u.,即为S1态的单点能。 将S1态的单点能与S0态的单点能相减,即得momap需要的两态的能量差参数Ead=0.07502 a.u.。
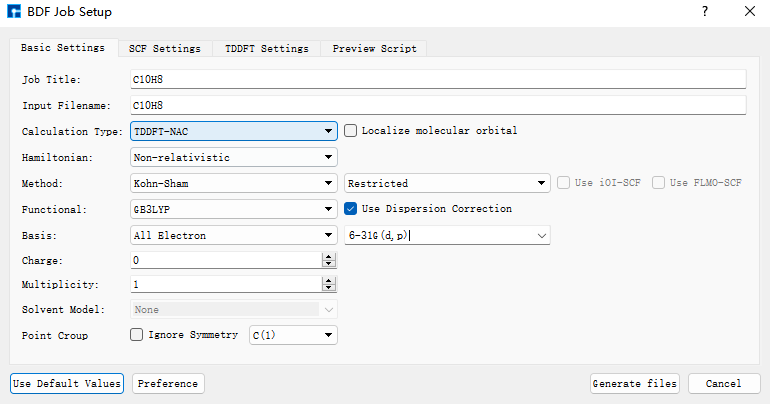
基于基态结构,做S0和S1之间的非绝热耦合计算。点击azulene_s0.hzw,右击点击copy,设置new file name为nacme,在Project中出现nacme.hzw。双击nacme.hzw,使用BDF进行azulene的非绝热耦合计算。选中Simulator → BDF → BDF,界面中设置参数。在Basic Settings界面中的Calculation Type选择TDDFT-NAC,方法采用默认的GB3LYP泛函,基组在Basis中的All Electron类型中,选择6-31G(d,p)。SCF Settings和TDDFT Settings面板中将Use MPEC+COSX Acceleraton的默认勾选去掉。在TDDFT Settings面板中的Non-Adiabatic Coupling内容框中,在默认的Coupling between Ground and Excited-State下,点击‘+’号,Basic Settings、SCF Settings、TDDFT Settings界面中的其它参数以及OPT Settings、Freq Settings等面板的参数使用推荐的默认值,不需要做修改。之后点击 Generate files 即可生成对应计算的输入文件。
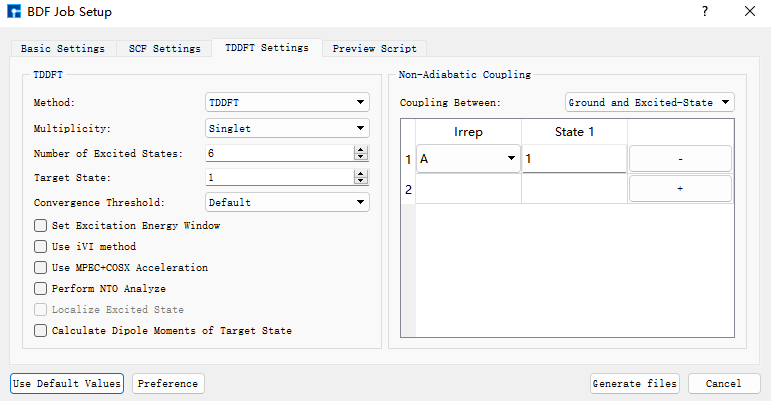
选中生成的bdf.inp文件,右击选择open with,打开该文件,如下所示:
$compass
Title
C10H8
Geometry
C 0.79273796 0.49102542 -0.00003306
C -0.70229648 0.61186591 0.00000000
C 1.30022931 1.80163336 -0.00006271
C -0.99262499 1.98726799 0.00007812
C 1.54415131 -0.67887417 0.00000000
C -1.63318173 -0.42094562 -0.00002837
C 0.21877157 2.69859812 0.00000000
C 1.10656346 -2.00562675 0.00005788
C -1.41619168 -1.80093044 -0.00004813
C -0.20258112 -2.49333482 0.00000000
H 2.35092512 2.06249888 -0.00009827
H -1.98777599 2.41348149 0.00017649
H 2.62424717 -0.53731744 0.00001117
H -2.67585843 -0.10561277 -0.00001520
H 0.30641472 3.77916915 0.00002385
H 1.88966565 -2.75951312 0.00017580
H -2.31053950 -2.41870504 -0.00009018
H -0.29054446 -3.57807510 0.00000000
End Geometry
Basis
6-31G(d,p)
Skeleton
Group
C(1)
$end
$xuanyuan
Direct
$end
$scf
RKS
Charge
0
SpinMulti
1
DFT
GB3LYP
D3
MPEC+COSX
Molden
$end
$tddft
Imethod
1
Isf
0
Idiag
1
Iroot
6
Istore
1
$end
$resp
Quad
FNAC
Norder
1
Method
2
Nfiles
1
Single
States
1
1 1 1
$end
选中bdf.inp文件,右击选择Run提交作业,任务结束后bdf.out,bdf.scf.molden三个结果文件会出现在Project中。 至此,MOMAP对azulene的S1→S0的辐射速率和内转换速率的计算需要的BDF量化软件的结构优化频率结果文件、非绝热耦合结果文件和参数部分都已完成。 下一期会接着介绍使用MOMAP进行azulene的S1→S0的辐射速率和内转换速率的计算。
-
ADC转换速率就是输出速率吗?2024-12-17 542
-
AD7192转换速率怎么算?2023-12-19 650
-
AD698转换速率慢怎么解决?2023-11-21 630
-
Azulene的S1→S0的荧光难以被观测到的原因2022-10-14 2699
-
如何使用MOMAP软件计算S1→S0的荧光辐射速率2022-09-15 6668
-
ACPI电源管理中的S0 S1 S2 S3 S4 S52022-01-06 2158
-
运算放大器的转换速率SR2019-06-11 5209
-
浅析转换速率SR2019-04-15 3057
-
请问AD9273是如何改变转换速率的?2018-10-09 2471
-
如何使用转换速率控制EMI2018-08-31 1462
-
恒定电压转换速率的设置方法2018-06-25 4297
-
ADE7755 S0 S1 SCF G0 G1接法问题2013-06-05 2681
-
转换速率,转换速率是什么意思2010-03-22 9267
全部0条评论

快来发表一下你的评论吧 !
