

原位电催化与红外、拉曼光谱联用
电子说
描述
前言
化学反应是生活生产必要的过程。化学反应的本质是旧化学键的断裂和新键的形成,但是在这个过程中,通常遇到两个问题:其一是反应能否直接进行,其二是结果是否为目标产物。当化学反应的动力学限制了以上两个问题时,我们可以使用催化剂加以解决。通常催化过程包括热催化、电催化、光催化等多种形式,其中电催化方式可以通过灵敏地控制条件从而控制反应进程。
通常电化学反应装置(图1)由三电极体系(参比电极、工作电极和对电极)和电解液组成,使用电化学工作站进行相关测量。在三电极体系中,使用电极电势相对稳定的参比电极提供标准的电势,作为基准,对电极只起到电流导通的作用,重要的研究过程在工作电极上发生。

图1 电化学反应池(图片来自网络)
电催化过程中电极上发生的反应是一个异相催化的氧化还原过程,反应在电极和液相的界面进行,因此在固液界面不断发生吸附与脱附、电荷转移、化学反应等过程。[1]在电极与液体的界面处,双电层间的场强达到108 V/cm,为稳定分子参与化学反应提供了条件。因此电化学的电极过程是电催化的重要过程。[2]然而,电化学测量过程只有电压-电流等信号之间的关系,电极材料的状态、电催化的过程无法得到全面认识。随着表征技术的发展,电镜、X射线衍射、红外、紫外-可见吸收光谱、拉曼光谱等多种手段应用到电极材料的表征中。催化过程是个动态过程,若只用上述手段测试始末状态,无法得到催化机理,因此电催化需要与其他表征技术联用,从而原位研究电催化的机理。在众多的技术中,扫描电镜、透射电镜等表征手段基于电子信号的收集,然而电子的平均自由程较短,此类的测试即使是原位电镜,也充满了难度(此外价格也是重要考虑因素之一)。因此,电催化的原位表征通常可以与红外、拉曼、原子力显微镜等技术联用。本文重点介绍红外与拉曼相关的工作。
原位红外
在光谱中,波长位于800 nm-1000 μm的电磁波称为红外光,红外光又分为近红外(800 nm-3μm)、中红外(3 -30 μm)以及远红外(大于30 μm)。由于中红外能量和分子的振转能级相当,可引起特征性的吸收,因此红外吸收光谱,通常指中红外部分。
红外光谱可以根据吸收峰进行对分子的定性分析,以及根据朗伯比尔定律进行定量分析,然而红外与电化学的联用却不是十分容易。表1列出常见的红外吸收频率与官能团。[3]由于电催化过程中水溶液是常见的反应体系,而水分子存在氢键,多个分子常常缔合在一起,因此水的吸收常表现为大于3300 cm-1 的吸收带。为红外光谱的测量带来麻烦。
|
表1 一些特征基团的振动频率[3] |
|||
|
特征基团 |
频率/cm-1 |
特征基团 |
频率/cm-1 |
|
OH伸缩 |
3800~3300 |
NH+ 4不对称变角 |
约1430 |
|
液体H2O对称、反对称伸缩 |
约3400 |
CO2- 3反对称伸缩 |
约1430 |
|
NH2反对称伸缩 |
3400~3300 |
COO-对称伸缩 |
约1410 |
|
NH2对称伸缩 |
3400~3200 |
CH3对称变角 |
约1375 |
|
炔类≡C-H伸缩 |
约3300 |
NO2对称伸缩 |
约1350 |
|
CH3反对称伸缩 |
约2960 |
NO-3反对称伸缩 |
约1350 |
|
CH2反对称伸缩 |
约2925 |
CH2扭曲振动 |
约1300 |
|
CH3对称伸缩 |
约2875 |
CH2面外摇摆 |
约1250 |
|
CH2对称伸缩 |
约2855 |
P═ O伸缩 |
约1250 |
|
═ CH伸缩 |
3100~3000 |
C-N伸缩 |
约1250 |
|
SH伸缩 |
约2550 |
S═O伸缩 |
约1250 |
|
CO2反对称伸缩 |
2380~2300 |
C-O-C(酯)反对称伸缩 |
约1200 |
|
C≡N伸缩 |
约2200 |
C-O-C(酯)对称伸缩 |
约1100 |
|
C═ O伸缩(酯、醛、酮、酸等) |
1760~1660 |
C-O-C(醚)反对称伸缩 |
约1100 |
|
H2O变角振动 |
1645 |
SO2- 4反对称伸缩 |
约1100 |
|
NH2变角振动 |
约1630 |
SiO2- 3反对称伸缩 |
约1100 |
|
NH+ 3不对称变角 |
1650~1600 |
PO3- 4反对称伸缩 |
1100~1050 |
|
C═ C伸缩 |
1640~1630 |
C-O伸缩 |
1100~1000 |
|
COO-反对称伸缩 |
1600~1550 |
P-O-C不对称伸缩 |
1060 |
|
NO2不对称伸缩 |
1550~1500 |
NO- 3对称伸缩 |
1050 |
|
NH+ 3对称变角 |
约1500 |
CHO(羧酸)面外弯曲 |
约930 |
|
CH2变角振动 |
约1465 |
CH2面内摇摆 |
730~720 |
|
CH3不对称变角 |
约1450 |
CO- 2剪式振动 |
约685 |
|
COH面内弯曲 |
约1430 |
CO- 2面外摇摆 |
约550 |
在1990年Moss等人[4]研究了透射原位电化学红外光谱的技术,图2为电化学原位池结构。由于常见的溴化钾材料可以溶于水,因此透光的窗口材料选择氟化钙,然而代价是损失部分光谱范围(氟化钙透光范围0.13~10 μm,溴化钾透光范围0.2~25μm)。该研究通过10 μm的光程,测量了10 mM马细胞色素在电催化过程中的红外信号变化。此类设计随后用于很多研究。[5-7]
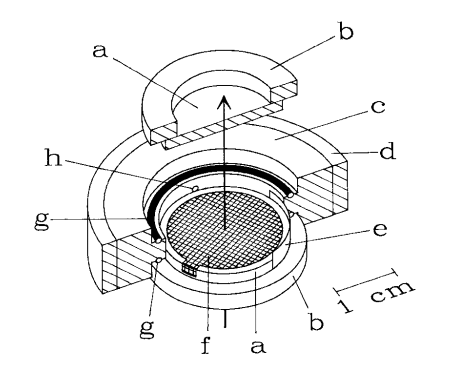
图2 透射原位电化学红外光谱的原位池结构示意图a CaF2窗口,b 有机玻璃环,c四氟乙烯池体,d钢外壳,e Pt对电极,f Au网工作电极g橡胶O圈h参比电极连接口[4]
随着红外技术的发展,反射式红外也可于电化学联用。利用衰减全反射方法可将非常薄的工作电极直接制作到衰减全反射晶体上,从而光不经过电解质,减少不必要的吸收损失,如图4所示。[8-10]
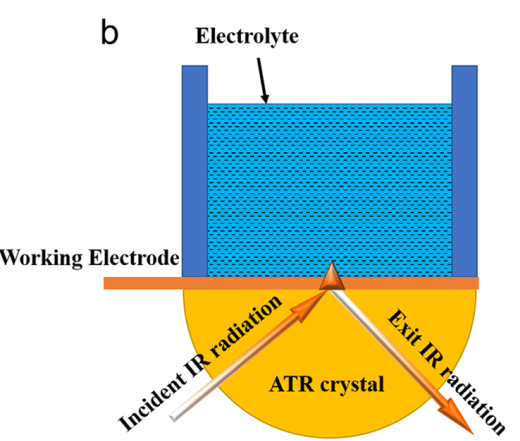
图3 衰减全反射原位电化学红外光谱原位池[8]
Mucalo[11, 12] 等设计了反射式原位电化学红外光谱样品池,如图4所示。红外光穿过窗片和电解质到达工作电极,然后反射出来,从而检测吸收光谱,如图3所示。此方式由于光程很长,因此光的衰减很多。
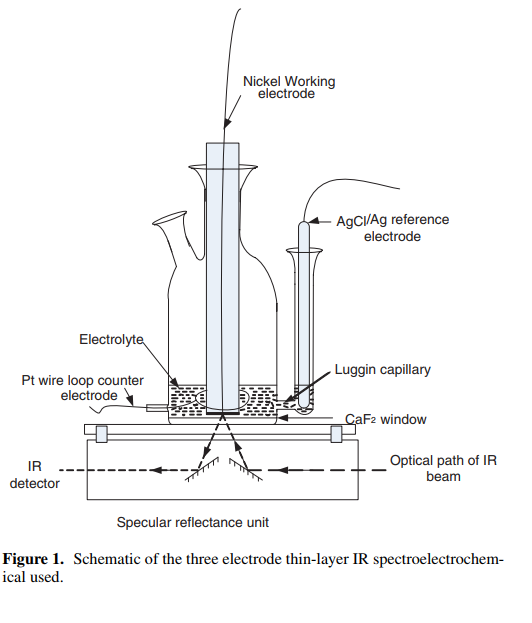
图4 三电极薄层红外光谱示意图
原位拉曼
拉曼光谱(Raman spectra),是一种散射光谱。拉曼光谱分析法是基于印度科学家C.V.拉曼(Raman)所发现的拉曼散射效应,对与入射光频率不同的散射光谱进行分析以得到分子振动、转动方面信息,并应用于分子结构研究的一种分析方法(参考百科)。
虽然拉曼光谱也是振动光谱,与红外技术相比,电化学原位拉曼过程则较少地受到光程、电解质吸附、窗片等因素的影响,搜索电化学原位拉曼会更容易得到原位池的结果[13],这是因为其光源在紫外、可见到近红外范围可变,在此范围内常见的窗片材料(如石英,蓝宝石等)和水等常见溶剂都吸收很弱,因此拉曼光谱的原位池构建和测量上相对方便很多,如图5所示,电化学与拉曼的联用池可以模块化,自由组合拆装。
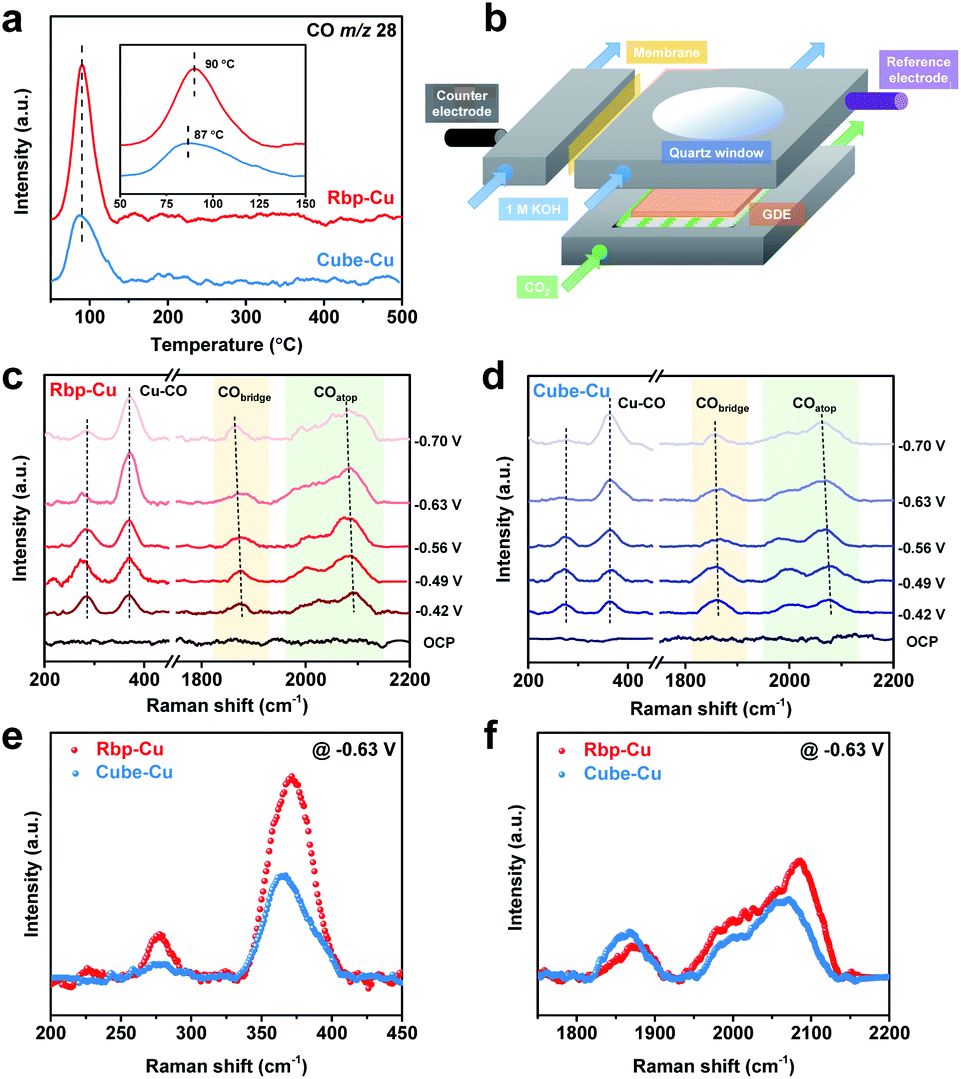
图5 原位拉曼与电化催化联用
存在问题
提到原位电化学的联用,通常是指在常温常压下使用红外、拉曼等手段表征电极上的化学反应过程,其他条件默认不计。但是这个“默认不计”却忽略了实验中两个重要的参数与电催化的关系,即温度和压力。通过能斯特方程可知,温度和压力是影响电极电势的两个重要因素,即使电势的0参考点—标准氢电极电势,也需要有温度和压力的限制。在有气体参与的过程中,反应器中的压力影响溶解平衡、吸附平衡进而影响电极电势,比如在氧还原的催化中,氧气的压力应当是大范围可控、可调的。
首先温度直接影响几乎所有过程。通过能斯特方程发现电极电势随温度变化;阿伦尼乌斯方程中温度改变化学反应速率;半导体催化剂中温度影响热激发载流子;测量中温度影响配分函数,从而在相同的背景下测量不同温度的红外光谱需要慎重分析。图6为不同温度条件下HER过程,温度可以很大程度上改变催化性质。[14]
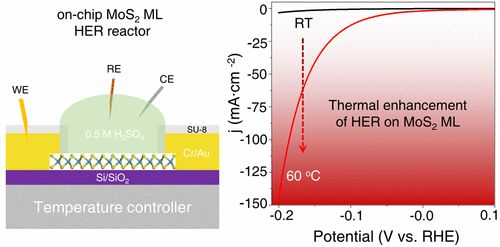
图6 温度影响HER过程
其次,气体参加的反应中压力影响溶解平衡、吸附平衡从而影响催化过程,甚至催化产物。在能斯特方程中,浓度的意义关联着平衡常数,而平衡常数则在对数运算中,因此这里的压力变化,需要解决气体密封到几倍大气压、十几个大气压、几十个大气压等量级的变化范围。耐压问题不仅仅涉及到原位池本身,参比电极等细小的组成部件的耐压问题同样需要考虑。因此在考虑以上两个因素后,电催化的联用技术中原位池的设计困难重重。不过正如前言所述,联用手段不同,面对的困难也有差别。
文献回顾
在考虑了温度和压力后,相同方向的工作可以提升到新的高度,下面罗列几篇相关报道。
Zenonas Jusys 和 R. Jürgen Behm [15]报道了电催化与原位红外联用进行甲醛氧化方面的工作,其中采集了不同温度、电压下的相关数据,并进行了相关动力学的分析。需要指出的是红外吸收光谱是测量谱和背景谱的差,而不同的温度不仅影响吸附脱附平衡,而且影响溶剂的状态,(甚至在更高的温度测量体系的红外辐射影响测量)因此不同温度下的背景往往不同,需要小心处理。
Olaf Brummel 和 Jörg Libuda 介绍了超高真空设备中模型催化剂的电化学与红外的联用(图7)。[16]通常我们使用的催化剂为粉末或多孔催化剂,此类催化剂有大量的边、角、楞暴露,而且具有丰富的点、线、面缺陷,因此催化性能高,但是我们在表征机理时由于难以区分吸附和催化转化的位点,因此得到的往往是各种位点性能的统计平均,无法进一步细化。研究人员研究单晶体系,由于可控的因素多(晶面、修饰、暴露量等)因此可以进行十分精细的研究。
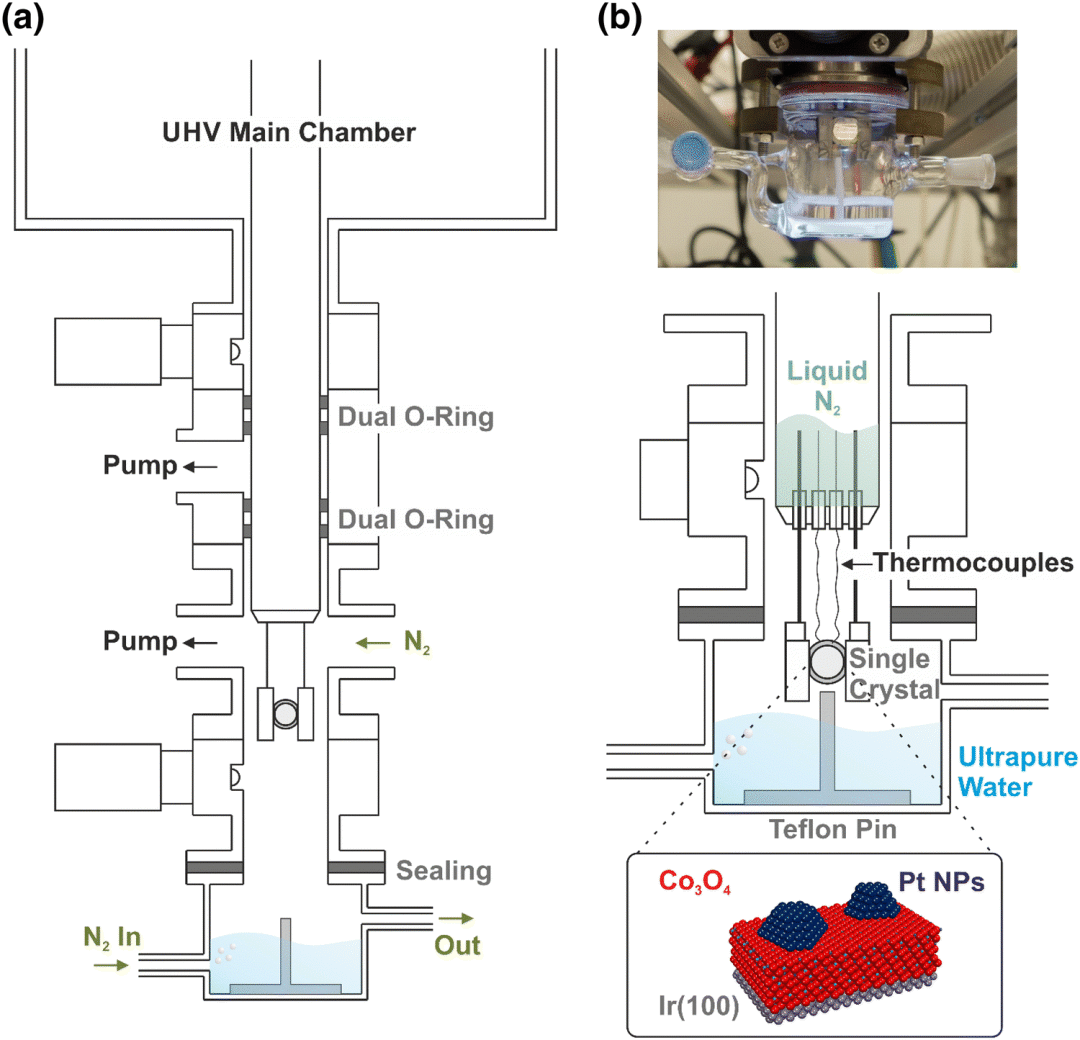
图7 a) Schematic representation of the UHV transfer system; b) sketch of the transfer process from UHV to the electrochemical environment[16]
WenChang Xie [17]使用原位拉曼研究了甲酸在Au@Pt表面电催化氧化过程中温度变化的影响(图8)。在这个体系中Au@Pt颗粒的粗糙表面也会有增强拉曼的效果,使得较弱的峰可以显现。然而增强拉曼却不是每个体系都存在的,只有在少数金属上拉曼信号才能有这个额外的加成。比如金属氧化物也是常见的工作电极,很多时候就只能依靠原始的拉曼信号。
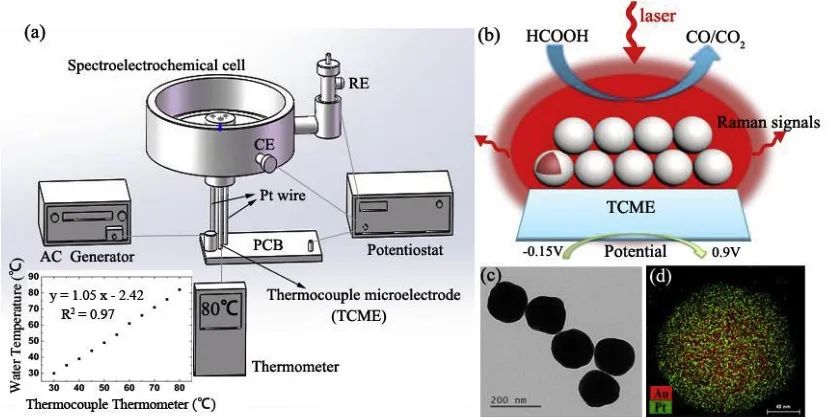
图8 (a) Device schematic diagram of high frequency heating technology combined with in situ spectroelectrochemical cell. Insert: Temperature calibration diagram of thermocouple microelectrode (b) Schematic diagram of in situ EC-SERS study of HCOOH electrooxidation on Au@Pt/TCME at elevated temperatures (c) TEM image of Au@Pt nanoparticles and (d) Element maps of the single particle of Au@Pt.[17]
需要注意的是荧光是干扰拉曼的重要信号,而很多共轭有机物、氧化物半导体等都有荧光,因此随着研究体系的不同,对激光器的要求也不同。为避免荧光的干扰,我们可以选用更长波长和更短波长的激光器,而短波长的激光有时对氧化物体系造成影响,如光生电荷分离和转移从而引起颗粒不同晶面的电势不同,与电催化外电路产生的电场相比,这个电场几乎不能忽略。相关文献可参考大连化物所范峰滔、李灿等人的工作(图9)[18]。因此在非金属材料强烈的激光照射下,是否存在自身的光敏性与双电层耦合,该过程是否进一步反馈给测量需要小心求证。
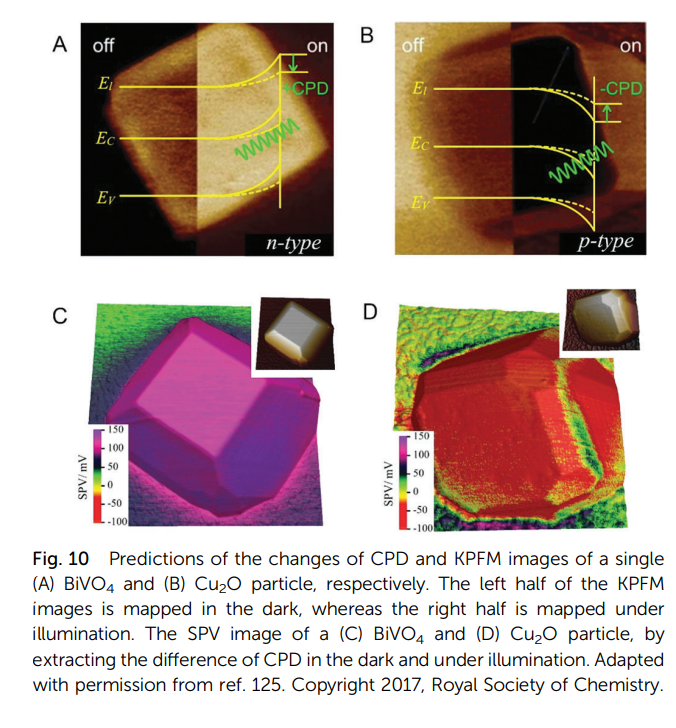
图9 BiVO4和Cu2O开尔文探针力显微图及表面光电压成像
小结与展望
写在最后,由于原位池的开发相关的工作需要一定的技术,也需要一定的资金,而且在课题中属于基础投资,存在见效时间和收益多少的问题,因此多自由度的原位池的研发不是那么丰富,甚至可以说是缺少,以此为基础的工作少之又少,举几个简单的例子,提供简单的思路。
电催化合成氨的文章能搜到许多,氮气氢气合成氨本身就是体积减小的反应,加压有利于平衡移动,但是几乎所有电催化合成氨的工作都是在常温常压下,搜不到加压条件下电催化合成氨的工作。本身氮气氢气难溶于水,已经不利于电极表面原料浓度的提高。而氨气极易溶于水,加压可以溶解更多,促进合成氨,可以使用流水将氨移除,进一步促进平衡转移,随后变压可以分离原料和产物(但是在气相中不易分离),优点很多,但是没有充分利用。(气相合成氨领域压力是重要的一个自由度,甚至不是热催化的工作都要考察,例如这篇机械力化学合成氨的nature[19],高压合成氨电化学方面有最新的工作Electrosynthesis of ammonia with high selectivity and high rates via engineering of the solid-electrolyte interphase,发表在《Joule》)。
前面提到了光电的效应,那么电催化中有没有其他的影响呢?比如热电效应、压电效应等。压电材料放到分离膜上,利用压力变化驱动材料,起到自清洁作用[13],那么压电材料作为电催化剂时,电化学池联合超声,是否能增强电催化呢?尚不可知。
总之由于电催化领域中体系改变的不多(一般是电催化材料,电解液和电压),反应条件(声、光、热、磁、电、力)改变的不多,自由度(温度、压力)控制不多,多变量下的联用测量也不多,使得电催化的空白很多。
审核编辑 :李倩
-
速读拉曼Raman光谱原理2019-07-13 0
-
拉曼Raman光谱与红外线光谱都是材质分析工具,那他们有什么差别呢?2019-07-13 0
-
共焦拉曼光谱仪的白光成像在生物分子拉曼测试中的应用2009-10-25 861
-
电催化与电催化电极的原理及研究2021-02-10 2820
-
重点介绍红外与拉曼相关的工作2022-09-05 2302
-
什么是拉曼光谱成像2023-04-10 1266
-
拉曼光谱技术系统详解2023-05-10 2600
-
原位拉曼系统--实时监测半导体薄膜生长全过程2023-08-14 922
-
拉曼光谱仪的原理及应用2023-09-09 10519
-
先进的拉曼光谱技术2024-01-15 369
-
探索拉曼光谱的奇妙世界:从原理到应用2024-06-12 564
-
TPIR 785 高通量高灵敏度拉曼光谱仪2024-06-26 320
-
拉曼光谱仪原理及应用2024-07-01 679
-
拉曼光谱的原理及其应用2024-08-26 380
全部0条评论

快来发表一下你的评论吧 !
