

利用海藻酸钠粘结剂和水溶剂制备CuF2电极可以抑制CuF2在有机电解质中的溶解
电子说
描述
背景介绍
锂离子电池中的转换型正极通常由Fe、Cu、O、S等成本较低且环境友好的元素组成,其容量比插层型正极大得多,过渡金属氟化物(MFx)既能提供》2.0 V的高氧化还原电位(由于金属-氟化物键的高离子性),又能在单位公式中允许多个电子转移而具有大容量,目前的主要挑战之一是其循环稳定性。氟化铜(CuF2)具有高理论电位(3.55 V vs. Li/Li+),高理论容量528 mAh/g,在所有金属氟化物正极中具有最高的能量密度。但由于在充电/放电循环期间严重的Cu 离子溶解,CuF2 正极循环不超过5圈。
二、正文部分
1、成果简介
近日,马里兰大学王春生教授,达尔豪斯大学杨崇银教授利用海藻酸钠(SA)粘结剂和水溶剂制备CuF2电极可以抑制CuF2在有机电解质中的溶解。使用水作为浆料溶剂和海藻酸钠 (SA) 作为粘合剂,在电极制造过程中在 CuF2 颗粒表面形成 2+ 配位海藻酸钠 (Cu-SA) 成功地抑制了铜的溶解。痕量溶解在水中的CuF2 中的2+ 可以与SA 粘合剂原位交联,在CuF2 表面形成保形Cu-SA 层。电极干燥过程中水分蒸发后,Cu-SA 层为锂离子导体但为 Cu2+ 绝缘体,可有效抑制 Cu 离子在有机 4 M LiCO4/EC-PC 电解质中的溶解,增强 CuF2 的可逆性。具有 SA 粘合剂的 CuF2 电极在 0.05 C 下循环 50 次后的可逆容量为 420.4 mAh/g,能量密度达到 1009.1 Wh kg-1证明了该策略的有效性。该研究以题目为“Super-reversible CuF2 cathodes enabled by Cu2+ coordinated alginate”的论文发表在国际顶级期刊《Advanced Materials》上。

2、研究亮点
本工作通过CuF2、水和SA混合到电极浆料实现在CuF2纳米粒子表面原位形成Cu2+配位SA层,交联的SA能够实现Li+的传输,但它阻碍Cu2+的传输,有效抑制CuF2正极中Cu离子在有机电解质中的溶解,减少电解质/电极界面之间的不良相互作用。实验结果显示, H-CuF2-SA电极在0.05C下循环50次,可逆容量420.4 mAh/g,能量密度1009Wh kg-1,高于大多数金属氟化物,体现了Cu基氟化物正极的优异性能。
3、图文导读
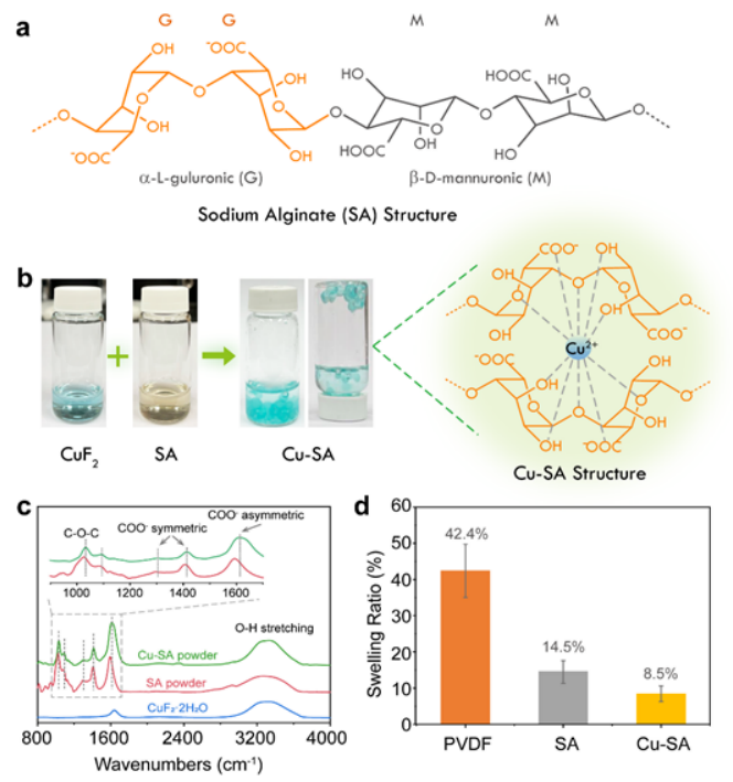
【图1】(a)海藻酸钠(SA)链的化学结构;(b) Cu-SA凝胶和Cu-SA化学结构的数字图像,直观展示SA和Cu2+离子的交联过程作用;(c)干燥后SA和Cu-SA粉末的拉曼光谱;(d) PVDF、SA和Cu-SA薄膜在EC/PC溶剂中的溶胀比。
SA是一种从褐藻中提取的天然多糖,是α- l -古洛醛基单元(G区)和β- d -甘露醛基单元(M区)的共聚物家族,含有大量的羟基和羧基(图1a)。这些G区中含有极性氧原子的富电官能团可以与多价阳离子交联,形成稳固的网络结构。将稀释后的CuF2水溶液逐滴加入SA水溶液中,SA分子立即与Cu2+离子发生相互作用,形成具有“蛋盒结构”的蓝色Cu-SA水凝胶(图1b)。利用傅里叶变换红外(FTIR)表征了SA与Cu2+离子之间的强交联。为了减少游离水的强-OH基团信号,在FTIR测量前,将Cu-SA水凝胶干燥并研磨成粉末。如图1c所示,SA粉末在~3300 cm-1处呈现出与氢键O-H伸缩振动相关的宽频吸收带,在1000 ~ 1700 cm-1处出现一系列与C-O键相关的尖锐峰与Cu2+交联后,C-O吸收峰发生明显的蓝移。 位于1592、1406和1026 cm-1的峰与COO-和C-O-C基团的不对称/对称振动有关,分别移至1608、1412和1032 cm-1。这些峰移为SA和Cu2+离子之间的化学相互作用提供了强有力的证据。
通过测定SA和Cu-SA膜在EC/PC有机溶剂中浸泡24 h后的增重,来评价SA和Cu-SA膜在EC/PC有机溶剂中较低的溶胀率,这对有效抑制Cu传输和快速锂离子传输是重要的,为比较PVDF膜在相同溶剂中的增重也进行了评价。如图1d所示,SA和Cu-SA膜的增重分别为14.5%和8.5%,而相同厚度的PVDF膜吸收了大量的碳酸盐溶剂,增重达42.4%。SA和Cu-SA的低膨胀性保证了Cu-SA层在CuF2表面的高稳定性。对水SA浆料制备的羟基化CuF2电极的电化学性能进行了评价,并与NMP-PVDF浆料制备的无水CuF2电极进行了比较。通过球磨CuF2-科琴黑(KB)混合物对无水纳米颗粒和羟基化CuF2进行碳包覆,以提高反应动力学。CuF2与科琴黑导电剂(KB)高能球磨(HBM)后,将不规则颗粒转化为平均粒径约为10 nm的CuF2纳米晶团块。
更重要的是,小的CuF2纳米颗粒被KB纳米颗粒连接并紧密包围,形成连续的导电网络,从而大大提高了CuF2的电子导电性。通过TEM对碳包覆的无水纳米颗粒和羟基化CuF2纳米颗粒的形貌进行了表征。
与原始样品不同的是,无水纳米颗粒在单个粒子中只能观察到一种类型的晶格条纹,而相应的SAED图像仍然以多个衍射环的形式显示,表明HBM CuF2纳米颗粒的单晶性。采用x射线粉末衍射(XRD)测定HBM处理后CuF2纳米颗粒的相结构。HBM处理后,无水CuF2样品没有观察到额外的峰,所有的峰与单斜CuF2 (JCPDS #42-1244)匹配良好,表明是无水CuF2的纯相。
然而,在HBM含水CuF2纳米颗粒中观察到与层状Cu(OH)F (JCPDS #07-0306)有关的新衍射峰,这是由于HBM过程中随着温度的升高部分含水CuF2分解。此外,HBM处理后明显拓宽的峰宽也证实了这些HBM CuF2样品的纳米晶性质。
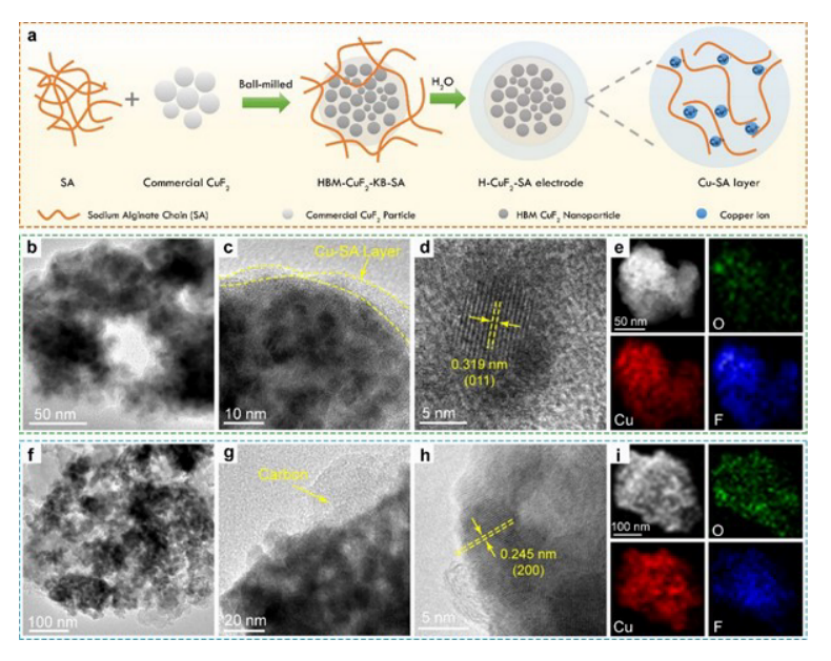
【图2】(a) H-CuF2-SA电极制备原理图;(b)(c)原始H-CuF2-SA电极纳米颗粒的TEM图像和(d) 原始H-CuF2-SA电极纳米颗粒的HRTEM图像;(e) H-CuF2-SA纳米颗粒的HAADF-STEM图像以及相应的Cu、F、O元素的EDS图谱;(f) (g)原始H - CuF2 -PVDF电极纳米颗粒的TEM图像和(h) HRTEM图像;(i) H-CuF2-PVDF电极纳米粒子的HAADF-STEM图像以及相应的Cu, F, O元素的EDS图谱。
图2a为SA粘结剂羟基化CuF2电极(简称H-CuF2-SA电极)的制备过程。与传统的电极制备工艺不同,我们没有直接将SA水溶液与CuF2颗粒混合,以避免SA与Cu2+离子之间的快速交联效应。我们先用CuF2纳米颗粒球磨SA粉,以保证SA分散均匀,与CuF2颗粒接触紧密。然后将KB加入到球磨的CuF2-SA混合物中,进一步球磨得到CuF2-KB-SA纳米颗粒(HBM-CuF2-KB-SA)。人工连续研磨过程中,滴加去离子水作为粘结剂溶剂到HBM-CuF2-KB-SA混合物中。CuF2溶解度低,SA溶解过程缓慢,可以有效地阻止大型Cu交联SA水凝胶(Cu-SA)的快速生长,确保在CuF2- kb表面原位形成薄的Cu-SA层。H-CuF2-SA电极粉末的TEM图像显示,CuF2表面的Cu-SA层厚度为~5 nm(图2b, 2c)。HRTEM图像显示Cu-SA层为非晶结构(图2c, 2d)。图2d中晶格平面间距为0.319 nm的纳米晶体可以被索引为Cu(OH)F的(011)晶格平面。由无水CuF2·2H2O到Cu(OH)F的相变是由于电极干燥过程中有水CuF2·2H2O的热分解引起的,这与相应的XRD图一致。Cu, F和O元素映射图也显示了Cu-SA和SA的均匀分布(图2e)。为进行对比,采用PVDF粘结剂和NMP溶剂制备了没有Cu-SA涂层的CuF2·2H2O-KB电极(简称H-CuF2-PVDF电极)。
由于H-CuF2-PVDF中的PVDF粘结剂很难观察到,其形貌与HBM-H-CuF2-KB纳米颗粒几乎相同(图2f-g)。平面间距为0.245 nm的单个粒子中的平行晶格条纹与Cu(OH)F的(200)晶格平面有关(图2h)。同时,HRTEM图像的特征圆晶格条纹为炭黑的特征条纹。
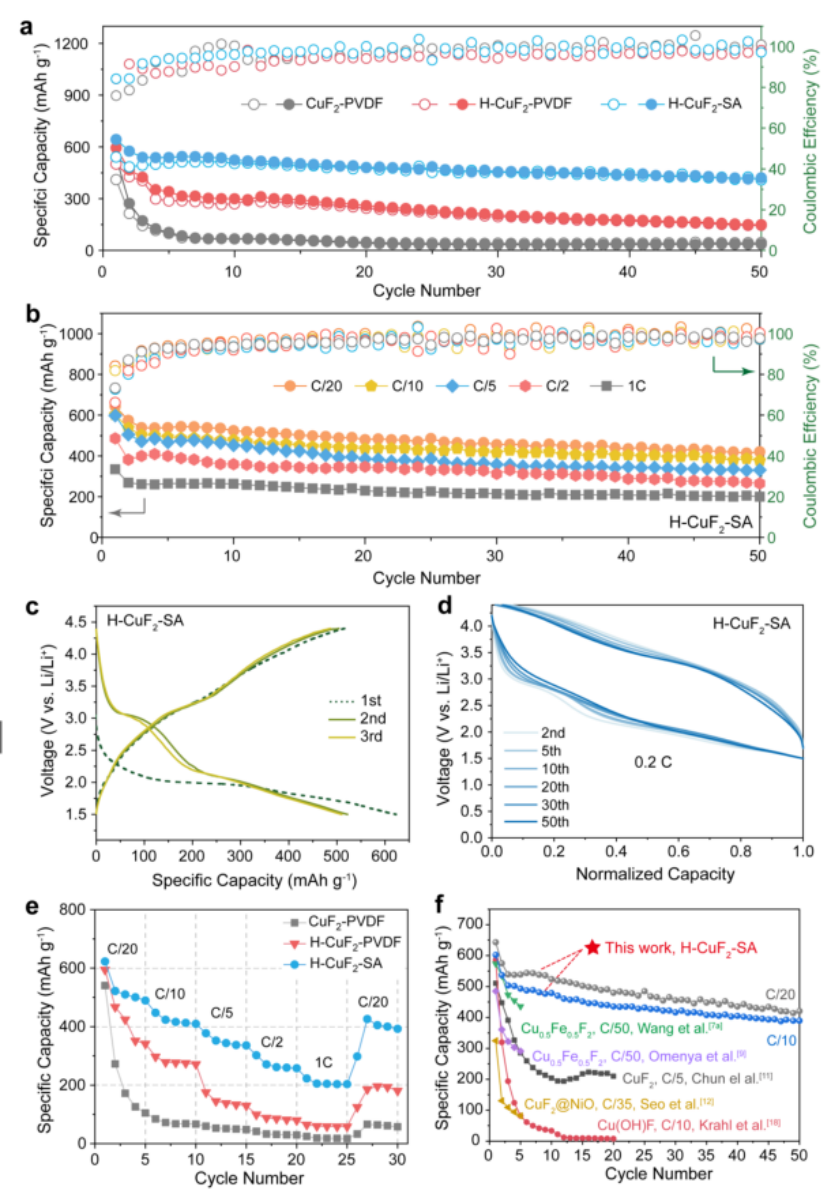
【图3】不同粘结剂对无水CuF2和羟基化CuF2正极的电化学性能研究。(a) 0.05 C下CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA正极的长循环性能;(b) H-CuF2-SA在不同倍率下的循环稳定性;(c) 0.05C(1C =539 mAh/g)时H-CuF2-SA的充放电曲线;(d) H-CuF2-SA电极在0.2C的归一化充放电曲线;(e) CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA正极的倍率性能;(f)与以往其他CuF2或铜基氟化物正极的循环性能比较。
图3a为0.05 c时H-CuF2-SA、H-CuF2-PVDF和CuF2-PVDF电极的循环性能,仅循环5次,CuF2-PVDF的容量迅速衰减至104.5 mAh/g,小于其初始容量的20%。与CuF2-PVDF相比,H-CuF2-PVDF具有更好的循环稳定性,但在50次循环时容量仍然下降到148.6 mAh/g。与之形成鲜明对比的是,H-CuF2-SA在0.05C下50次循环时保持了420.4 mAh/g的可逆容量,这在过去的报道中从未有过。我们还评估了三种电极在不同倍率下的长周期稳定性(图3b)。CuF2-PVDF和H-CuF2-PVDF在所有倍率下的循环稳定性都很差。而H-CuF2-SA(图3f)在0.05C时能达到420.4 mAh/g的容量,在1C的高速率下循环50次后能保持200.1 mAh/g的容量,是CuF2-PVDF或H-CuF2-PVDF的5-10倍。
采用恒流充放电法研究了H-CuF2-SA、H-CuF2-PVDF和CuF2-PVDF电极在0.05C下的电化学性能。CuF2-PVDF在第一次放电中平坦的平台在2.9V左右来自于CuF2的一步还原反应,形成金属铜和氟化锂在0.05C下,CuF2-PVDF的首次放电容量为540.2 mAh/g,高于理论容量528 mAh/g,这是由于电解液与电极之间的寄生反应所致。在第一次装药过程中,出现两个斜坡平台3.4V和3.7V分别于与Cu(0)在3.4V时氧化生成Cu(I)中间体,以及Cu(0)和Cu(I)在3.7V时同时氧化生成Cu(II)产物有关。3.7V的高电位氧化平台主要来源于对CuF2的再转换。但由于3.5V的热力学势相似,铜被电化学氧化成铜离子以及铜离子溶解到电解液中也对铜的容量有贡献。407.8 mAh/g的低首次荷电容量主要是Cu颗粒与LiF基体反应不完全和Cu离子溶解所致。由于带电过程中活性物质的溶解是不可逆的,CuF2正极的二次放电容量降至272.5 mAh/g。CuF2-PVDF在3.1V第二次放电中的变化归因于颗粒尺寸的减小导致反应动力学的增加。CuF2-PVDF由于铜溶解严重,在前3次循环后放电容量迅速下降至31.8%。
与CuF2-PVDF不同的是,H-CuF2-SA和H-CuF2-PVDF的第一恒流放电曲线在2.0 V附近出现了一个漫长的平台期,在0.05 C时分别达到了622.8和588.3 mAh/g的高比容量(图3c)。Cu-OH的电负性比Cu-F低,降低了氟化铜转化的活化能垒,从而降低了还原电位。在以下电荷分布中,H-CuF2-PVDF和H-CuF2-SA在3.2V处都有一个小的平台和大斜坡平台在3.7V。与CuF2-PVDF的负极反应相似,在3.2V处出现的小平台。V是铜金属氧化为Cu(I)中间体的产物,3.7V处斜坡平台较大是由于Cu(OH)F的生成。在第二次及后续放电中,H-CuF2-SA和H-CuF2-PVDF电极在3.1V和2.1V附近均表现出较高的放电电位。由于H-CuF2-PVDF在充电过程中 Cu离子在电解液中的溶解,导致放电容量迅速衰减。值得一提的是,由于LiF形成的能垒较大,在初始循环到3.1V容量的衰减较快。
由于Cu-SA涂层抑制了Cu阳离子溶解,H-CuF2-SA表现出稳定的容量(图3c)。H-CuF2-SA的可逆转换由归一化容量的充放电曲线进一步证明(图3d)。循环50次后,H-CuF2-SA的还原电位在3.0 V和2.1V。而H-CuF2-PVDF和CuF2-PVDF的电池循环20次后,在3.0 V的还原反应几乎消失。
在1.5 ~ 4V的电压范围内,采用0.1 mV/s的扫描速率下的循环伏安法(CV)评价了CuF2-PVDF、H-CuF2-SA和H-CuF2-PVDF电极的电化学行为。三种氟电极均在3.0 V左右出现Cu-F的锂化反应峰,而H-CuF2-PVDF和H-CuF2-SA均出现在2.0 V的较低电位处有额外尖锐的锂化反应峰,对应CuF2羟基化部分的锂化。与无水CuF2不同,水合氟化物在消耗过程中在3.0 V以下有两个额外的小峰。H-CuF2-PVDF和H-CuF2-SA也具有较低的氧化峰3.4V(CuF2-PVDF的是3.7V)。这些结果表明LiOH的反应活化能垒比LiF低,这与它们的恒电流分布相一致。三电极的CV曲线与三电极的恒流充放电行为一致。
此外,H-CuF2-SA也表现出比H-CuF2-PVDF和CuF2-PVDF更优异的倍率 (图3e)。在0.05C的低速率下,三种电极的首次放电容量相近,约为600 mAh/g。当电流密度增加到0.1、0.2、0.5和1 C时,H-CuF2-SA的可逆容量分别为409.4、335.8、257.8和203.3 mAh/g。当电流密度恢复到0.05 C时,电池具有稳定的可逆放电容量392.5 mAh/g,表现出良好的循环稳定性和速率耐受性。而在相同速率下,H-CuF2-PVDF的容量仅为341.5、271.8、129.7、79.7和58.1 mAh/g,在改变倍率前,CuF2-PVDF的容量以0.05C的低速率在5个循环中迅速衰减到《100 mAh/g。
图3f比较了迄今为止报道的基于CuF2的转换正极。所有报道的基于CuF2的转换正极在前5个周期内迅速下降,无论在0.2C的高倍率还是在0.02C的极低倍率下。与之形成鲜明对比的是,H-CuF2-SA可以实现50次可逆循环,并保持420.4 mAh/g的容量,在所有的CuF2基正极中表现出优越的循环稳定性。在H-CuF2电极制造过程中,原位交联Cu-SA层结构简单,对提高循环稳定性非常有效。
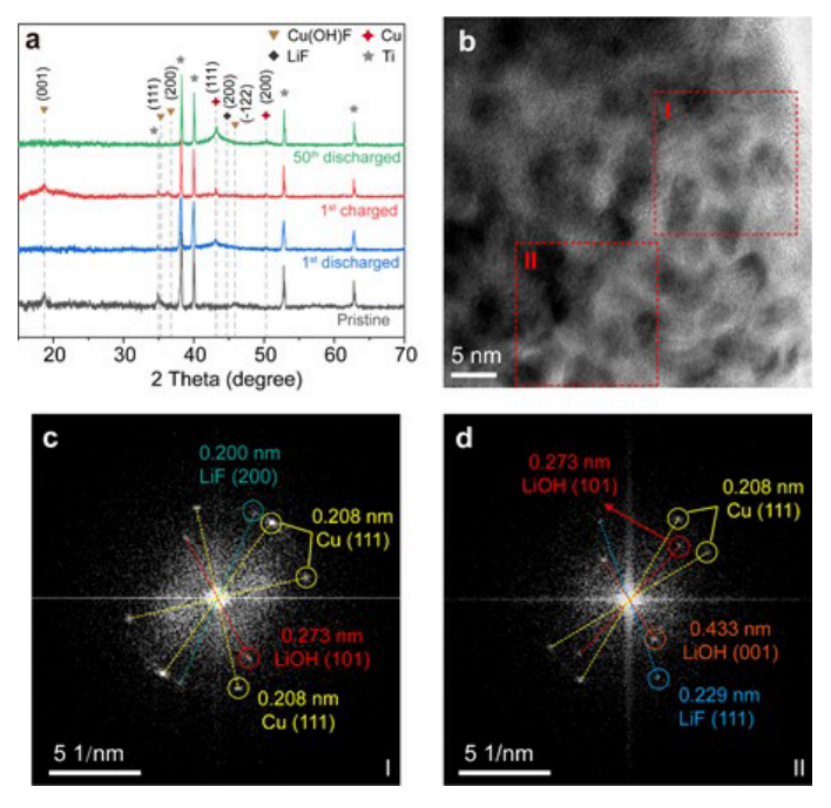
【图4】(a)不同充放电状态下H-CuF2-SA正极的XRD谱图;(b) H-CuF2-SA正极第一次放电后的HRTEM图像;(c)区域I和(d)区域II对应的FFT模式。
通过XRD研究了H-CuF2-SA的相变和反应可逆性(图4a)。当H-CuF2-SA完全放电至1.5V。Cu(OH)F在(001)面附近18.7°处的峰消失,而在43.2°附近Cu(111)面出现了一个明显的宽峰,说明Cu(OH)F发生了充分的转化。值得注意的是,由于LiF和LiOH的结晶度较低,且与Cu和Ti的峰位重叠,所以在第一放电态时看不到其他峰。当电池充满电时,Cu(OH)F的峰值恢复,表明Cu(OH)F的可逆反应。但仍能发现铜的一个小而尖的峰,表明有大量非活性铜颗粒留在电极中的痕迹。循环50次后,Cu的衍射峰由较大的宽峰和较小的尖峰组成,说明Cu在长周期循环后颗粒尺寸没有严重变粗,这保证了H-CuF2-SA电极良好的循环性能。
HRTEM图像和对应的FFT谱图进一步证明了LiF和LiOH的存在(图4b-d)。由于Cu、LiF和LiOH纳米颗粒的叠加分布,使得它们在HRTEM图像中的晶格面重叠,难以分辨(图4b)。本文选取HRTEM图像中的两个区域(图4b中的红框)进行快速傅里叶变换(FFT)分析,以明确LiF和LiOH的存在。I区域对应的FFT图显示了三组衍射斑,表明了该区域的多晶分布(图4c)。晶格面距离为0.208 nm的最亮衍射点与高度结晶的Cu(111)面相关,这与XRD结果一致。除此之外,较暗且晶格距离较低的点可以索引到LiF的(200)平面。由于(111)平面是Cu的最大平面距离,因此(111)平面距离较大的衍射斑很容易与Cu区分开来。因此在区域I和区域II中,位于较小圆上的衍射点可以根据特征晶格距离分别索引到LiF的(111)平面和LiOH的(101),(001)平面(图4c, d)。
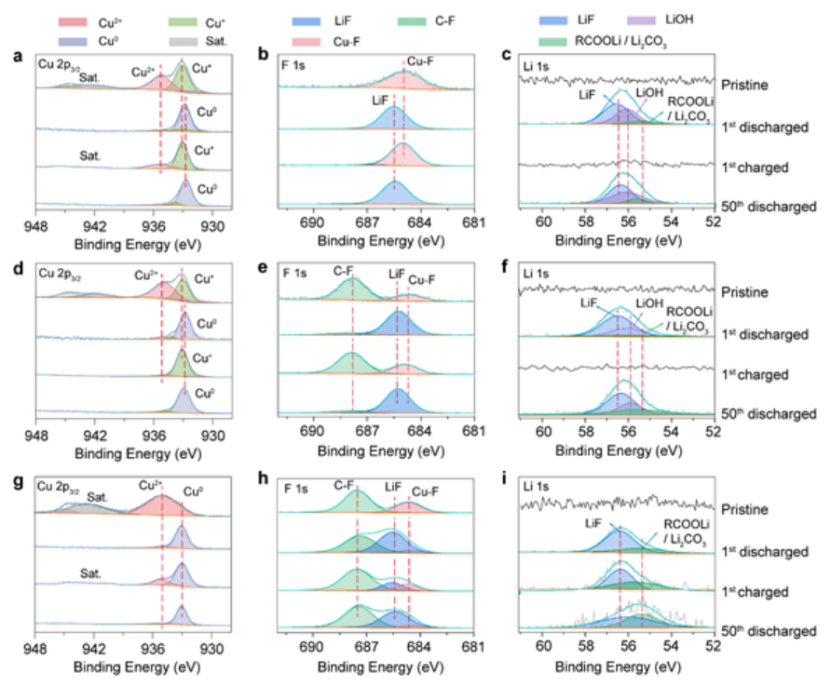
【图5】(a-c) H-CuF2-SA (d-f) H-CuF2-PVDF和(g-i) CuF2-PVDF电极在循环前后的Cu 2p2/3、F 1s和Li 1s的XPS谱。每个光谱都是从电极表面进行测试的。
对原始和循环CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA电极进行XPS分析揭示了它们的可逆性(图5)。对于两个水合的原始H-CuF2-SA(图5a)和H-CuF2-PVDF(图5d)电极,Cu 2p3/2的光谱可以很好地模拟成两个峰,分别位于933.1 eV和935.2 eV,这可以归因于Cu+和Cu2+氧化态,表明铜在Cu(OH)F中的多价性而原始CuF2-PVDF电极中仅观察到一个位于935.7 eV的与Cu2+-F相关的单峰(图5g)。当电池第一次放电到1.5 V时,3个电极的Cu 2p3/2光谱移至932.6 eV,索引为金属态的Cu0,表明这些氟化电极完全还原。在第一次完全充电状态下,原始样品中的Cu2+和Cu+价态在H-CuF2-SA电极中恢复,表明其转化反应具有良好的可逆性。而H-CuF2-PVDF中,由于电极表面电荷过程中铜溶解严重,Cu2+信号几乎消失,这与其充放电性能相符合。在无水CuF2- PVDF电极上,Cu0峰的存在表明,由于Cu颗粒的粗化只有部分金属铜重新转化为CuF2。在F1s光谱中也观察到了类似的结果。原始电极中的Cu-F信号在放电状态下完全转化为Li-F信号,在H-CuF2-PVDF和H-CuF2-SA电极中完全充电状态下恢复,而在不同的嵌锂态下,CuF2-PVDF电极中都能检测到Li-F和Cu-F信号。纯CuF2不完全转化的主要原因是导电金属Cu与隔离的LiF聚集体之间的电子传输中断,导致CuF2- PVDF电极中存在大量“死”活性物质。放电态的Li 1s光谱揭示了纯CuF2和羟基化CuF2之间的不同转换反应路径(图5c, 5f, 5i)。在放电状态下,H-CuF2-PVDF和H-CuF2-SA电极的f1s光谱中都能检测到Li-F和Li-OH,这有力地证明了还原反应中形成了LiF和LiOH的混合物,而不是纯LiF。LiOH的存在有效地降低了转换反应的自由焓,从而保证了Cu(OH)F的反应比纯CuF2正极材料更彻底。
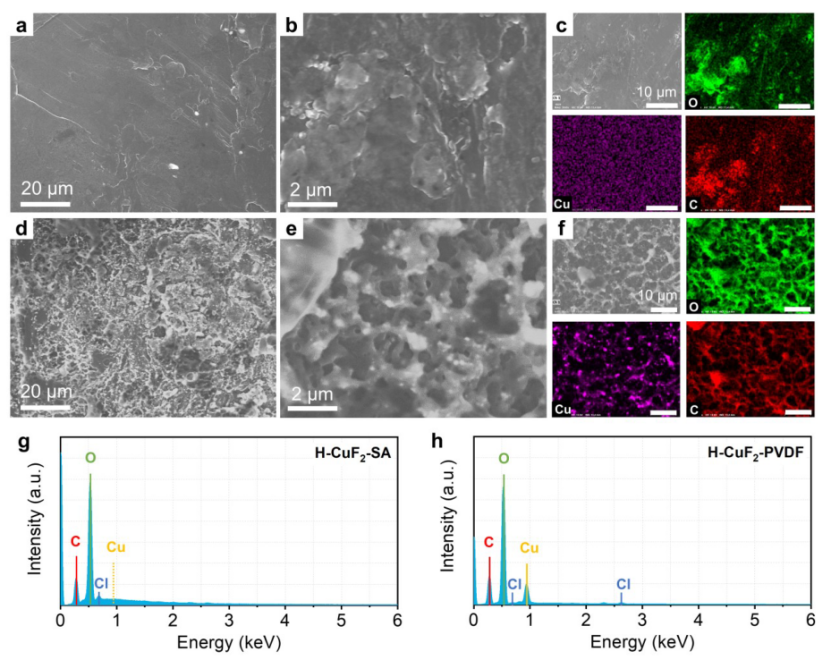
【图6】在与Li负极搭配循环50次后的(a-c)H-CuF2-SA正极;(d-f) H-CuF2-PVDF正极的SEM图和相应的Cu、O、C元素分布图。(g) H-CuF2-SA和(h) H-CuF2-PVDF正极循环后Li负极表面的EDS光谱图。
用透射电镜(TEM)表征了Cu- SA涂层50次循环后Cu颗粒的生长和稳定性。金属Cu在H-CuF2-PVDF电极上经过50次循环后的还原产物在5 ~ 50 nm范围内尺寸不均匀。相比之下,H-CuF2-SA中的Cu颗粒仍保持均匀分布,平均粒径约为5 nm,这与上述XRD结果一致。循环50次后,Cu-SA层仍然清晰可见,作为预制CEI层,有助于提高H-CuF2-SA电极在长时间循环过程中的结构稳定性。
由于溶解于电解液中Cu离子会沉积在Li负极上,这可以通过SEM和EDS分析。如图6a-b所示,循环的Li负极与H-CuF2-SA正极配合后,表面光滑,仅可见少量盐渣,图6c所示的C和O元素分布图也进一步证明。同时,如图6c所示的Cu元素映射图像中出现了随机分布的点,颜色为淡蓝色,这可以归因于背景噪声。在相应的EDS光谱中未观察到Cu信号的痕迹(图6g),说明H-CuF2-SA正极在锂化/蚀除过程中没有发生明显的铜溶解。与之形成鲜明对比的是,Li负极与H-CuF2-PVDF正极循环后的SEM图像显示,Li负极表面极其粗糙,在Li表面镀有大量的Cu颗粒(图6d-e)。Cu元素映射图中出现明亮的蓝色斑点,对应的EDS光谱中出现较强的Cu峰,进一步表明Cu含量显著(图6f, 6h),说明H-CuF2-PVDF正极中铜溶解严重。此外,由于Li负极表面粗糙与Cu金属共沉淀有关,可以认为Cu离子的减少破坏了SEI在Li负极上的均匀性,导致表面粗糙。研究了锂化过渡金属正极/石墨电池充放电过程中过渡金属在SEI中对石墨的溶解和处置。 这里,CuF2电极中铜的溶解更为严重。
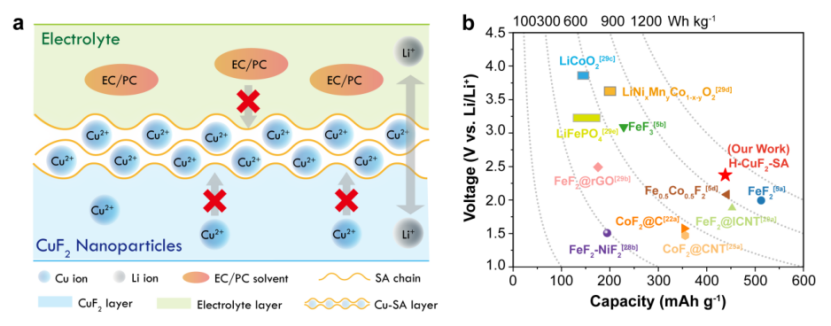
【图7】(a) SA粘结剂增强H-CuF2-SA正极电化学性能的工作机理示意图;(b)与其他金属氟化物和插层正极相比,H-CuF2-SA的容量、电压和能量密度(放电能量密度根据0.05 C的放电电流计算)。
对H-CuF2-SA正极的综合表征和电化学分析表明,H-CuF2-SA正极的超循环稳定性归因于电极制备过程中原位形成的独特的Cu-SA涂层。图7a示意图说明了H-CuF2颗粒上Cu-SA涂层的形成和作用。在电极制备过程中,由于溶液中SA链与铜离子之间的强交联作用,在CuF2颗粒表面原位形成了坚固的Cu-SA层。Cu-SA层对Li+选择性渗透,对Cu2+不渗透,有效地抑制了Cu离子的溶解和降解。此外,Cu-SA层和SA粘结剂在碳酸盐电解质溶剂中的溶胀性较低,降低了电极/电解液的相互作用,防止了电解液以不良的方式进入粘结剂/电极界面,从而进一步降低了铜在电极中溶解的可能性。此外,Cu-SA涂层交联到具有高刚性的基体中,保持了H-CuF2-SA电极在锂化/蚀除过程中的结构稳定性,确保H-CuF2-SA电极具有优异的稳定性,具有优越的可逆容量。虽然水基SA粘结剂将水化杂质引入CuF2正极材料中,H-CuF2-SA正极仍能提供2.4V以上的平均放电电位。高容量与高放电电压相结合,循环50次,电流密度为0.05 C时可产生约1009 Wh kg-1的可逆放电能量密度 (图7b),高于大多数转换金属氟化物,如FeF3、Fe2、Ni2和Co2。更重要的是,本研究中H-CuF2-SA的能量密度高于大多数铁基氟化物和商业插层正极材料。铜基氟正极的循环稳定性从5次显著提高到50次,表明其从不可逆向可逆的突破。
4、总结与展望
本工作通过在CuF2纳米粒子表面原位形成Cu2+配位SA层,成功抑制了CuF2正极中Cu离子的溶解。Cu-SA交联层在碳酸盐岩电解质中具有较低的溶胀性和较高的刚性,可以选择性地促进Li离子的转运,同时阻断Cu离子的迁移,减少电解质/电极界面之间的不良相互作用。制备的H-CuF2-SA电极在0.05C下循环50次,可逆容量420.4 mAh/g,能量密度1009Wh kg-1。优良的循环稳定性和能量密度证明了铜基氟化物正极的最佳性能。铜离子交联策略为CuF2作为下一代锂离子电池的高能、低成本和可逆正极打开了大门。
审核编辑 :李倩
-
锂离子电池电解液有机溶剂的发展趋势2013-06-17 5947
-
锂离子电池电解液超全面介绍 有何神秘之处?2017-02-22 7293
-
柔性印制电路中粘结剂的典型应用2018-09-10 2158
-
粘结剂在柔性印制电路中是如何使用的?2021-04-26 1892
-
金属锂表面预处理和电解液添加剂对锂电极表面的改性介绍2017-10-11 1320
-
阐述硅基负极材料粘结剂的研究进展并对不同类型粘结剂进行优缺点对比2018-02-01 20625
-
锂电池CMC粘结剂的四大特点、性能作用及挑选2020-04-03 58613
-
电池电解液和电解质的区别_电池电解液和电解质的两种形态2020-04-16 25719
-
宁德时代公开“一种固态电解质的制备方法”专利2021-01-20 4173
-
利用微流控芯片制备海藻酸钠微球2022-04-12 5793
-
氟化铜(CuF2)在金属氟化物正极中的作用2022-09-20 5962
-
一文读懂锂电粘结剂现状及技术发展2023-12-11 4224
-
干法 vs 湿法工艺:全固态锂电池复合正极中粘结剂分布与电荷传输机制2025-08-11 2204
全部0条评论

快来发表一下你的评论吧 !
