

揭示浓缩WiS电解质中EDL的详细原子结构
电子说
描述
背景介绍
盐包水电解质因其安全性和低毒性而成为未来电化学储能装置的一个有吸引力的选择。 然而,在电极和盐包水电解质之间的界面处发生的物理化学相互作用尚未完全了解。
正文部分
1、成果简介
美国埃默里大学Lian Tianquan教授和华中科技大学冯光教授等人通过原位拉曼光谱和分子动力学模拟,研究了在盐包水电解质和 Au(111) 电极之间的界面处发生的双电层结构。作者证明大多数界面水分子与锂离子结合并具有零个、一个或两个氢键。此外,锂离子在大的负极化下在电极表面的积累会降低界面场,从而导致界面水的不寻常的“H-up”结构和羟基拉伸频率的蓝移。
这种对双电层结构的原子解析为设计用于电化学储能装置的未来水系电解质提供了关键见解。该研究以题目为“Unconventional interfacial water structure of highly concentrated aqueous electrolytes at negative electrode polarizations”的论文发表在国际顶级期刊《Nature Communications》。

2、研究亮点
该工作揭示了浓缩盐包水电解质中双电层的详细原子结构,为在分子水平上理解高浓度条件下的界面锂离子行为提供了重要的见解,这将有利于 WiS 电解质系统中的电极表面工程。
3、图文导读
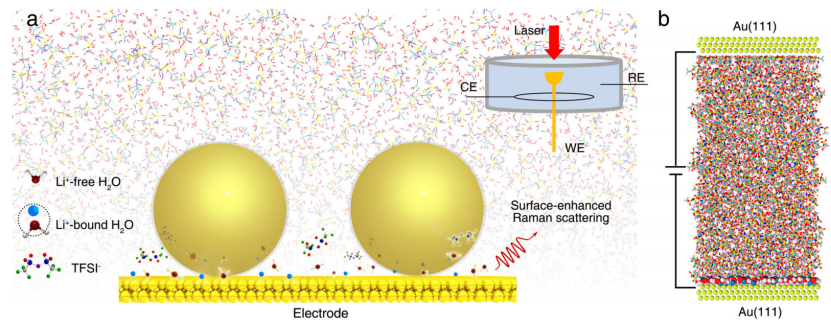
【图1】探索电极界面处的电化学双电层结构。a 使用 SHINES 方法对 EDL 进行原位探测的示意图。左下:Li+游离水、Li+结合水分子和TFSI-阴离子的结构示意图; 右上:光谱电化学电池示意图,其中 Au(111) 电极、Pt 线和 Ag/AgCl 电极分别用作工作电极 (WE)、对电极 (CE) 和参比电极 (RE)。 底部中心的大球是核/壳 Au/SiO2 纳米球。 b 在施加偏压下与 Au(111) 电极接触的 21 m WiS 电解质的典型 MD 模拟。电极表面的界面水分子被放大。
如图 1a 所示,作者为了研究双电层 (EDL) 结构,使用了电化学 SHINES 方法。该方法已被证明适用于电化学界面的研究,例如硫酸根离子、吡啶和氢的特异性吸附;最重要的是,能够在单晶电极表面上对双电层和界面水的结构进行原位分子水平探测。如图 1b 中的示意图所示,MD 模拟用于研究 EDL 的原子结构和 Au(111) 电极-盐包水(WiS)电解质界面处的水氢键结构。
作者通过原位电化学拉曼光谱在 21 m LiTFSI 水系电解质中测量了 OH 拉伸区域中界面水的振动光谱及其随施加电位的变化。所有电极电位均与 Au(111) 电极的零电荷电位(PZC) 相关,约为 -0.1 V vs. Ag/AgCl 电极。图 2a 是从 +0.5 到 -1.55 V条件下界面水 OH 拉伸模式在 3200-3600 cm-1 区域的拉曼光谱。
受低浓度水电解质中水的拉曼光谱分配的启发,该光谱可以很好地由三个高斯峰 1、2 和 3(随着频率增加)的总和拟合,表明EDL中的三种主要类型的水分子。如图 2b、c 所示,这三个峰的频率和强度都表现出对施加电位的强烈依赖性,反映了界面水结构和电场的偏置依赖性变化。峰 1-3 的拉曼频率从 +0.1 线性下降到 -1.15 V,这与Stark效应引起的频移一致,这是由于界面水在 0 到 -0.96 V 的电位区域所经历的总电场单调增加引起的 ,如图 2d 中的 MD 模拟所揭示的。MD模拟揭示了界面水在高负电位区域所经历的总电场从-0.96到-1.51 V的降低(图2d),这解释了水OH拉伸模式中的蓝移。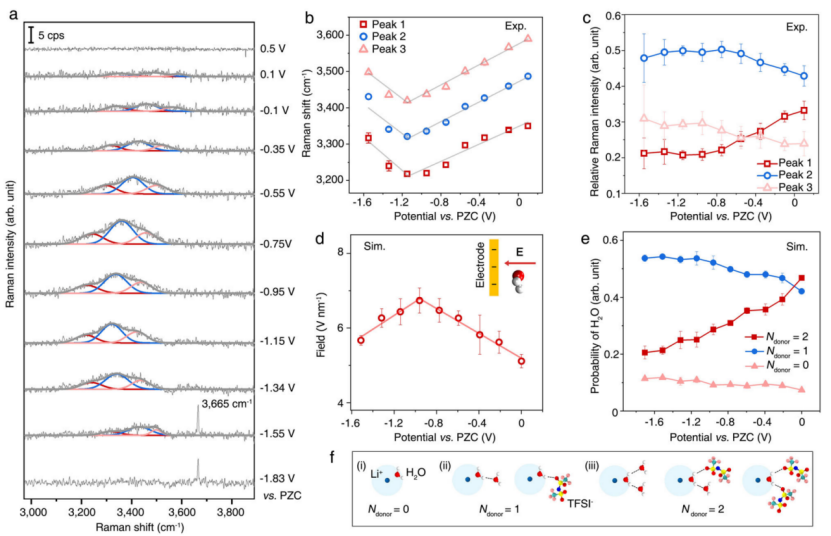
【图2】Au(111)|WiS 电解质 (21 m LiTFSI) 界面处界面水的振动拉曼光谱。a 在 21 m LiTFSI 水性电解质中测量的 Au(111) 表面界面水的 OH 拉伸模式的原位电化学拉曼光谱(灰色曲线)及其与三个频率增加的高斯峰之和的拟合:峰 1(红色)、2(蓝色)和 3(粉红色)。 b 在+0.1 ~−1.15 V和-1.15 ~−1.55 V区域,通过光谱拟合和线性拟合(实线)得到界面水的峰1(红色方框)、峰2(蓝色圆圈)和峰3(粉色三角形)的电位相关频率。
c 从 a 中的拟合获得的峰 1(红色方块)、2(蓝色圆圈)和 3(粉红色三角形)的相对强度的潜在依赖性。在此,使用代表总强度部分变化的相对拉曼强度分布来避免电位扫描期间等离子体增强的变化。 d 0~-0.96 V和-0.96~-1.51 V区域内界面水和线性拟合(红色实线)经历的模拟电场(E)强度(红色圆圈)。插图:电场和界面水之间相互作用的示意图。
E的方向被认为是它施加在带正电粒子上的力的方向,例如,从正极到负极。 e 模拟界面水的电位相关概率,给体数分别为 0(粉红色三角形)、1(蓝色圆圈)和 2(红色方块)。b-e 中的误差条表示实验或模拟中的标准误差。 f 给体数分别从 (i) 0 变为 (ii) 1 和 (iii) 2 的 Li+ 结合界面水示意图。在以 Li+ 结合的水分子作为给体形成 H 键时,另一个水分子或 TFSI- 可以作为受体。
氢键数的变化改变了 EDL 的结构,并且可以通过拉曼频移观察到,因为 OH 拉伸模式 (vOH) 的频率向具有较低氢键数的水分子中的较高区域移动。具有零受体数的 Li+ 结合水分子在界面区域中占主导地位,因为界面 Li+ 结合水分子及其 O 原子与 Li+ 相互作用(示意图显示在图 1a 的左侧)不能用作H键受体。
因此,如图 2e、f 所示,界面 Li+ 结合水分子的 H 键环境的主要差异取决于它们的 H 键给体数(Ndonor),其范围为 Ndonor = 0、1 和 2。如图2e所示,Ndonor = 2的界面水分子比例逐渐降低,Ndonor = 1和0的水分子比例在更负电位下相应增加。基于在稀电解质溶液中建立的 Ndonor 和 vOH 之间的关系,界面拉曼光谱的峰 1、2 和 3(按波数递增的顺序)分别归因于具有两个(Ndonor = 2),一个(Ndonor = 1)和零个(Ndonor = 0)氢键。
这些物种的模拟电位依赖性概率(图 2e)与观察到的其相对拉曼强度的趋势(图 2c)定性一致,为这一分配提供了进一步的支持。尽管结果表明,稀溶液中 Ndonor 和水的 vOH 之间已确立的关系也可以应用于浓缩电解质,但这一概念应在未来的研究中进一步研究。
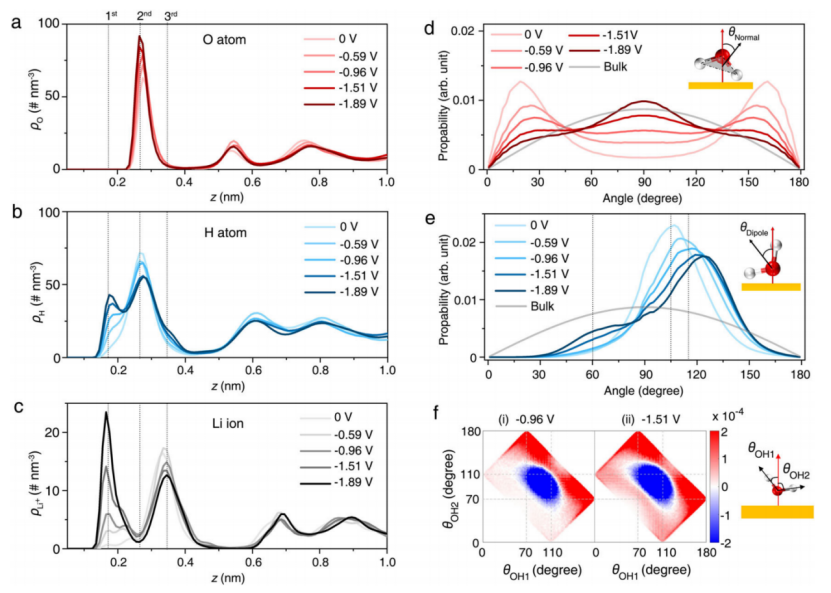
【图3】Au(111)|WiS电解质(21 m LiTFSI)界面处双电层的微观结构。a-c 水的氧 (a) 和氢 (b) 以及 21 m LiTFSI 水性电解质中的 Li+ (c)的原子数密度 (ρ)。 d,e,界面水的正常取向(d)和偶极取向(e)。法线方向定义为水平面法线与电极表面法线的夹角;偶极方向是水矢量与电极表面法线之间的夹角。灰色实线代表块状水的方向;e 中的三条虚线分别表示 (i) 105°、(ii) 115° 和 (iii) 60° 处的峰值。f (i) -0.96 V 和 (ii) -1.51 V 处 Li+ 结合水的两个 OH 基团相对于 PZC 下水的排列的差分二维角分布。 θOH 定义为水的 OH 键与电极表面法线方向的夹角(对应示意图如右图所示)。在 d-f 中,水分子由白色和红色球体表示。
MD 模拟揭示了 WiS 电解质 EDL 中界面水、Li+ 和 TFSI- 的详细原子结构。21 m LiTFSI 水系电解质中界面水和 Li+ 的结构可以通过界面水的氧(图 3a)和氢(图 3b)原子和 Li+(图 3c)的密度分布的数量,以及水的取向(图3d,e)来可视化。两者都显示出对施加电位的明显依赖性。水相对于电极表面的方向可以用两个角度来描述:1)水平面的法线向量与电极表面的夹角(θnormal);2)水偶极子和表面法线向量之间的角度(θdipole),如图3e所示。同时,Li+结合水的两个OH基团相对于PZC下水的排列的差分二维角分布如图3f所示。
水的氢和氧原子以及 Li+ 的数密度分布在距电极三个距离处表现出明显的峰,表明它们的界面结构顺序。具体而言,观察到位于第二层的氧原子(距表面约 0.26 nm)有一个尖峰,并且在更负的电势下,峰高增加,其位置更靠近电极表面(图 3a)。然而,如图 3b 所示,氢原子的分布表现出更多的电位依赖性变化。在 PZC 处,氢原子与氧原子位于同一层,θnormal在 20 和 160o 附近表现出两个峰值(图 3d),θdipole分布在 105o 处达到峰值(图 3e),这表明界面水采用几乎平行于电极表面的结构。随着极化增加到~-1.0 V,水分子明显重新排列:H原子分布在第一层显示出一个新峰(到表面~0.17 nm),第二层峰高降低( 图 3a、b);θnormal的分布变得不那么有序(图 3d)并且θdipole分布的峰值转移到 115o(图 3e)。
这些变化可归因于重新定向的“偶极向下”结构。然而,在高极化下,作者观察到 H 原子肩峰在第三层出现(到表面约 0.35 nm,见图 3b),并且在 60o 左右的θdipole分布的峰值逐渐增加(图 3e)。这种结构可以归因于一种不寻常的“偶极结构”。为了更准确地描述界面 Li+ 结合水分子在施加电位下的结构转变,作者计算了 Li+ 结合水的两个 OH 基团相对于 PZC 下水的排列的微分二维角分布。具体来说,水分子的 OH 键可分为“H-up”、“平行”和“H-down”三种。 如图 3f 所示,Li+ 结合水分子的排列在低极化下由平行调整为“H-down”。然而,在高极化下,虽然主要的 Li+ 结合水分子从平行转移到“H-down”,但部分水分子转移到不寻常的 H-up 构型。
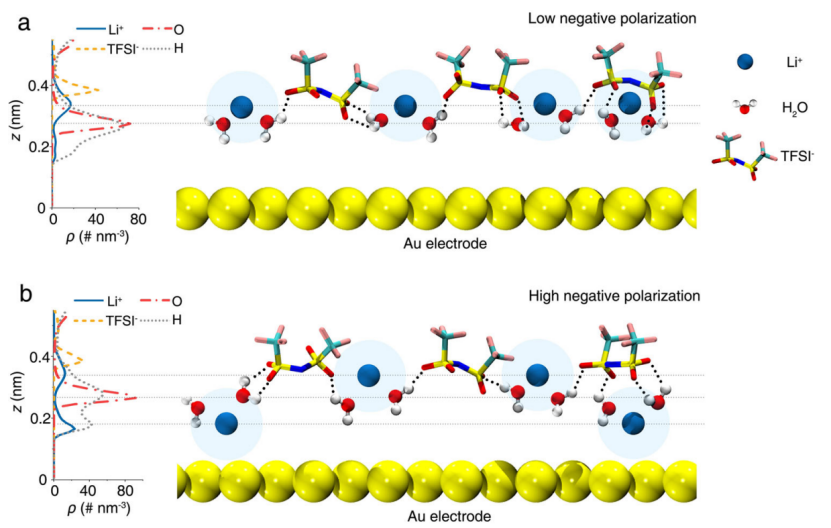
【图4】高浓度水系电解质中的原子 EDL 结构示意图。a, b 各种界面物质的数密度分布(左),以及在低负极化(从 0 到 ~-0.6 V)和高负极化(从 ~-1.5 到 -1.9)下相应的示意性 EDL 结构(右)。 水平虚线表示水的 Li+、O 和 H 原子密度分布的峰值以及 TFSI- 的质心。在 -1.55 V 时在 Au(111) 表面沉积 LiOH被省略。
结合原位电化学拉曼光谱和恒电位 MD 模拟结果的分析,作者提出了高浓度水系电解质中的 EDL 模型。如图 4 所示,大多数界面水分子位于 Li+ 的第一个溶剂化壳内,它们的 O 原子与 Li+ 相互作用,它们的 H 原子形成 0、1 和 2 个 H 键,其中 TFSI- 作为主要 H-键合受体。双电层中的电解质在沿电极表面法线的密度分布中显示出结构顺序:最接近电极表面的水分子与上方的 Li+ 和 TFSI- 离子层相互作用。
在低极化(从0到~-0.6 V)下,界面水分子采用平行和H-down结构(图4a)。然而,在高极化(从~-1.5 到 -1.9 V)下,锂离子在电极表面积累,插入电极和水层之间(图 4b)。结果,采用“H-up”取向的界面Li+结合水的量从~10%(PZC)增加到~20%(高极化)。
如图2b,d所示,具有不同H键数的界面水分子的电位依赖性OH伸缩频移与界面水所经历的平均电场密切相关,表明Stark效应引起的频移。为了了解电位相关的双电层结构变化如何导致观察到的频移,作者将界面水(红色圆圈)上的总电场分解为来自电极(H2O-电极,灰色条)的、水分子(H2O-H2O,橙色条)的以及 Li+ 和 TFSI- 离子(H2O-LiTFSI,蓝色条)的,如图 5a 所示。发现在 21 m的电解质中,由于表面电荷密度单调增加(图 5b),来自电极的电场强度随施加的电势线性增加(图 5a)。类似地,指向相反方向的水分子的场强也随电位单调增加(图5a中的橙色条)。
因此,总电场的异常转变主要是由 Li+ 和 TFSI- 离子的贡献引起的。如图 3c、5c所示,有两层锂离子:界面水上方的外层产生与带负电的电极方向相同的电场,电极和界面水层之间的内层产生反向电场(参见图 5d 中的示意图)。Li+ 层的峰值位置几乎与极化无关。外层锂离子的量从0到-0.96 V略有增加,然后从-0.96到-1.51 V下降更明显;虽然内层中的锂离子量在整个电位范围内增加,但增加的斜率从-0.96到-1.51 V要大得多(图5c)。外层和内层之间的锂离子数量差异(△ρ)在0到-0.96 V的电位逐渐减小,然后在-0.96到-1.51 V的电位区域逐渐减小。
因此,因此,Li+在电极表面积累,这屏蔽了负极表面电荷,在~-1.0 V的总电场的非常规转变中起主导作用(图5a)。
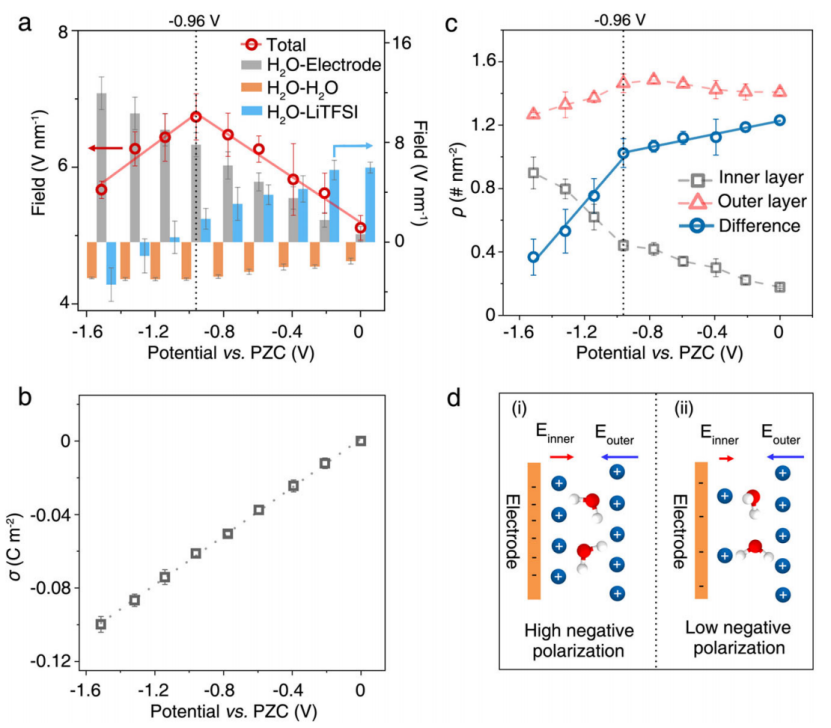
【图5】对电场变化的分子洞察。a 界面水所经历的模拟电场强度作为外加电位的函数,从 0 到 -1.51 V。总电场(红色圆圈)线性拟合(红色实线),如左轴所示。这样的总电场分别分解为 H2O 电极(灰色条)、H2O-H2O(橙色条)和 H2O-LiTFSI 电解质(蓝色条)相互作用,为清楚起见,它们在右轴上重新缩放。 b 表面电荷密度 (σ) 作为电位的函数。c 内层(灰色方块)和外层(红色三角形)中锂离子的电位依赖累积数密度(ρ),以及外层和内层之间离子数密度的相应差异(蓝色圆圈)。在 a-c 中,误差线表示标准误差。d 内层 (i) 和外层 (ii) 中锂离子诱导和产生的界面水所经历的电场示意图。外层的锂离子产生与带负电的电极产生方向相同的电场;而内层产生反向电场。水分子由白色和红色球体表示。
总之,通过结合原位振动光谱和恒电位 MD 模拟,作者研究了在 Au(111) 电极上高浓度 (21 m LiTFSI) 水系电解质的 EDL 的原子结构。界面水 OH 拉伸模式的拉曼光谱显示三个峰,其相对强度和频率取决于电位。MD 模拟表明 >93% 的界面水分子位于 Li+ 的第一个溶剂化壳中。这些界面水分子不能作为 H 键受体而是给体,因为它们的 O 原子与 Li+ 配位。它们的 H 键环境在 H 键给体数上有所不同,范围为 0、1 和 2,这与观察到的三个频率降低的 OH 伸缩峰很好地对应。这种分配得到了观察到的这些峰的相对拉曼强度的潜在依赖性,以及与水分子与这些给体数的模拟概率之间的良好一致性的支持。
由于Stark效应引起的振动频移,所有三个 OH峰的频率在从 PZC 到 -1.15 V 的高负电极极化处移至较低值;而它们表现出从-1.15到-1.55 V的蓝移,这在低浓度电解质中没有观察到。MD 模拟表明,在这个电位范围内,WiS 电解质中的锂离子会在电极表面积累,插入电极和第一层水分子之间。这降低了界面水分子所经历的电场强度,导致观察到其 OH 伸缩频率的蓝移。它还导致不寻常的“H-up”界面水分子,尽管有负极极化,但偶极子指向远离电极表面。最后,在更负的电位下,观察到电解质的分解反应在 Au(111) 表面产生 LiOH 沉积物。
4、总结和展望
该工作揭示了浓缩 WiS 电解质中 EDL 的详细原子结构,并确定了在高负极化下与低浓度电解质不同的独特结构特征。这些发现为在分子水平上理解高浓度条件下的界面锂离子行为提供了重要的见解,这将有利于 WiS 电解质系统中的电极表面工程。
审核编辑:刘清
-
无极电容器有电解质吗,无极电容器电解质怎么测2024-10-01 1503
-
不同类型的电池的电解质都是什么?2024-02-27 3523
-
Science综述:设计更好的电解质2022-12-13 1503
-
钠离子电池的电解质分类2022-10-09 6299
-
电池电解液和电解质的区别_电池电解液和电解质的两种形态2020-04-16 25399
-
聚蠕虫状聚电解质刷的吸附2019-10-24 1280
-
超薄电解质电容器问世 手机可迎袖珍化时代2014-09-24 3232
-
我想自己测试电解质2013-03-09 2711
-
电解质的作用是什么?2009-11-09 3968
-
电池内的电解质是什么?2009-10-20 1161
-
原子结构2009-08-06 2164
-
铁的原子结构示意图2008-05-28 124063
全部0条评论

快来发表一下你的评论吧 !
