

氟化铜(CuF2)在金属氟化物正极中的作用
电子说
描述
01 导读
由于其高理论电位(3.55 V)和高容量(528 mAh/g),氟化铜(CuF2)在所有金属氟化物正极中具有最高的能量密度。然而,CuF2只能进行不到5个循环,这主要是由于在充电/放电循环期间Cu离子严重溶解。
02 成果背景
近日,马里兰大学王春生教授和戴尔豪西大学杨崇英(助理)教授等在电极制造过程中使用水作为浆料溶剂和海藻酸钠 (SA) 作为粘合剂,通过在CuF2颗粒表面形成Cu2+配位的海藻酸钠(Cu-SA),成功抑制了铜溶解。在0.05 C下循环50次后,具有SA粘合剂的CuF2电极的可逆容量为420.4 mAh g-1,能量密度达到1009.1 Wh kg-1。相关工作以“Super-reversible CuF2 Cathodes Enabled by Cu2+ Coordinated Alginate”为题发表在Advanced Materials上。
03 关键创新
作者利用在CuF2纳米颗粒表面原位形成Cu2+配位SA层的简单策略,成功地抑制了CuF2正极中Cu离子的溶解,实现了高可逆CuF2正极。
04 核心内容解读
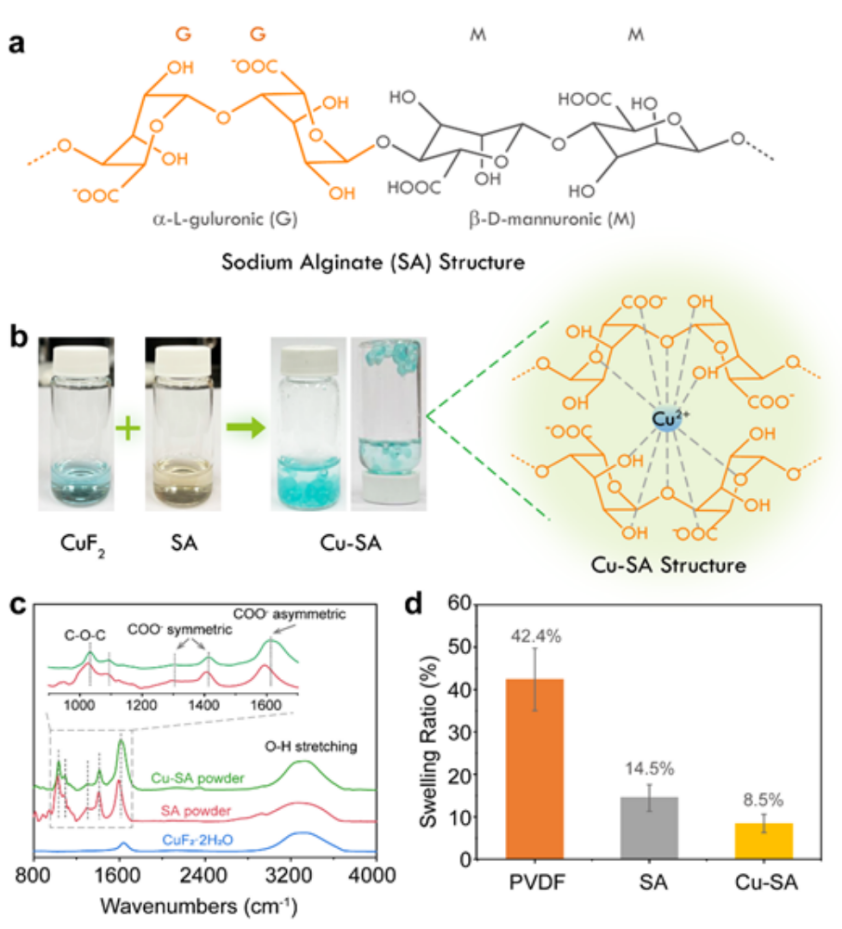
图1 (a)海藻酸钠(SA)链的化学结构;(b)Cu-SA凝胶的照片和Cu-SA的化学结构,直观地演示了SA和Cu2+离子交联作用。干燥的SA和Cu-SA粉末的(c)拉曼光谱;(d)PVDF、SA和Cu-SA薄膜在EC/PC溶剂中的膨胀比。@The Authors
作为一种从褐藻中提取的天然多糖,SA包含大量的羟基和羧基(图1a)。将稀释的CuF2水溶液滴加入SA水溶液中,SA分子立即与Cu2+离子相互作用,形成一个具有“蛋盒结构”的蓝色Cu-SA水凝胶(图1b)。
作者利用傅里叶变换红外(FTIR)表征了SA和Cu2+离子之间的强交联。如图1c所示,SA粉末在~3300cm-1处表现出氢键键合的O-H伸缩振动相关的宽吸收带,在1000~1700cm-1范围内有一系列对应的尖锐峰。与Cu2+离子交联后,C-O吸收峰出现明显的蓝移。位于1592、1406和1026 cm-1处的峰与COO-的不对称/对称振动和C-O-C基团的不对称振动有关,分别移至1608、1412和1032 cm-1。这些峰的位移为SA和Cu2+离子之间的化学相互作用提供了强有力的证据。
SA和Cu-SA薄膜在EC/PC有机溶剂中的溶胀性较小,这对有效抑制Cu传输和实现快速锂离子传输具有重要意义。如图1d所示,在EC/PC有机溶剂浸泡24个小时,SA和Cu-SA膜的重量分别增加了14.5%和8.5%,而厚度相近的PVDF膜吸收了大量的碳酸盐溶剂,重量增加达到42.4%。SA和Cu-SA的低膨胀度,保证了Cu-SA层在CuF2表面的高稳定性。
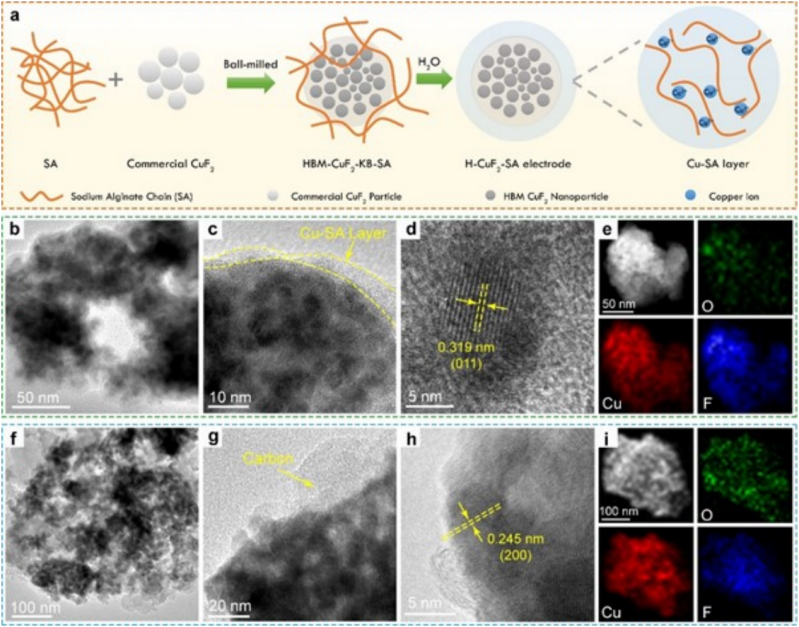
图2 (a)H-CuF2-SA电极的制备示意图;原始H-CuF2-SA电极纳米颗粒的(b)(c)TEM图像和(d)HRTEM图像;(e)H-CuF2-SA纳米颗粒的HAADF-STEM图像,及Cu、F、O元素的EDS;H-CuF2-PVDF电极纳米颗粒的(f)(g)TEM和(h) HRTEM图像;(i)H-CuF2-PVDF电极纳米颗粒的HAADF-STEM图像及相应的Cu、F、O元素的EDS。@The Authors
图2a展示了用SA粘合剂的羟基化CuF2电极(表示为H-CuF2-SA电极)的制备过程。与传统的电极制备工艺不同,作者没有直接将SA水溶液与CuF2粒子混合,以避免SA和Cu2+离子之间的快速交联效应。相反,作者首先用CuF2纳米颗粒球磨SA粉,以确保SA的均匀分散,并与CuF2颗粒紧密接触。然后作者将KB加入球磨的CuF2-SA混合物中,进一步球磨得到CuF2-KB-SA纳米颗粒(HBM-CuF2-KB-SA)。
H-CuF2-SA电极粉末的TEM图像显示,CuF2表面的Cu-SA层的厚度为~5 nm(图2b,2c)。从HRTEM图像中可以看出,Cu-SA层具有无定形结构(图2c,2d)。Cu、F和O的元素能谱图显示了Cu-SA和SA的均匀分布(图2e)。为了进行比较,作者还采用PVDF粘合剂和NMP溶剂制备了不含Cu-SA涂层的CuF2·2H2O-KB电极(表示为H-CuF2-PVDF电极)。
由于H-CuF2-PVDF中的PVDF粘合剂很难被观察到,其形态与HBM-H-CuF2-KB纳米颗粒几乎相同(图2f-g)。平面间距为0.245 nm的单个粒子中的平行晶格条纹与Cu(OH)F的(200)晶格平面有关(图2h)。同时,HRTEM图像上的特征圆形晶格条纹是炭黑的特征条纹。
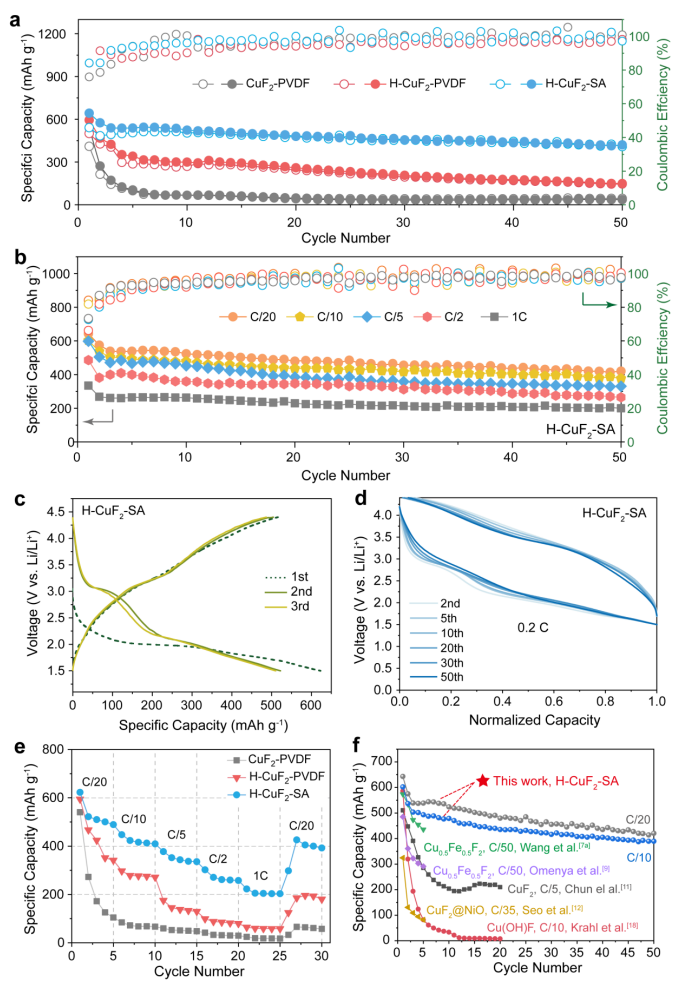
图3不同粘合剂对无水CuF2和羟基化CuF2正极的电化学性能。(a) 在0.05 C下,CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA正极的长循环性能;(b)在不同倍率下,H-CuF2-SA的循环稳定性;(c)H-CuF2-SA在0.05 C(1 C=519 mAh g-1)的充放电曲线;(d)0.2C下,H-CuF2-SA电极的充放电曲线;(e)CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA正极的倍率性能;(f)与其他CuF2或铜基氟化物正极的循环性能比较。@The Authors
图3a显示了H-CuF2-SA、H-CuF2-PVDF和CuF2-PVDF电极在0.05 C下的循环性能。仅经过5个循环后,CuF2-PVDF的容量迅速下降到104.5 mAh g-1,低于其初始容量的20%。H-CuF2-PVDF比CuF2-PVDF具有更好的循环稳定性,但在50次循环时,其容量仍下降到148.6 mAh g-1。与之形成鲜明对比的是,H-CuF2-SA在0.05 C下进行50个循环时保持了420.4 mAh g-1的可逆容量,这是以前从未报道过的。
作者还评估了三种电极在不同倍率下的长循环稳定性(图3b)。H-CuF2-SA(图3f)在0.05 C时,可达到420.4 mAh g-1的容量,在1 C高倍率下50个循环后保持200.1 mAh g-1,是CuF2-PVDF或H-CuF2-PVDF的5-10倍。与CuF2-PVDF不同,在0.05 C下,H-CuF2-SA和H-CuF2-PVDF的第一次恒流放电曲线显示出一个在2.0 V左右的长平台,分别达到622.8和588.3 mAh g-1的高比容量(图3c)。还原电位较低是由于Cu-OH的电负性低于Cu-F,降低了氟化铜转化的活化能垒。在第二次及之后的放电中,H-CuF2-SA和H-CuF2-PVDF电极在3.1 V和2.1 V左右均具有较高的放电电位。
由于充电过程中电解质中铜离子的溶解,H-CuF2-PVDF的放电容量迅速衰减。值得一提的是,由于氟化锂形成的能量势垒更大,在3.1 V时的容量在初始循环中衰减得更快。由于Cu-SA涂层抑制了Cu阳离子的溶解,H-CuF2-SA显示出稳定的容量(图3c)。通过归一化容量的充放电曲线进一步证明了H-CuF2-SA的可逆转换(图3d)。经过50个循环后,H-CuF2-SA在3.0 V和2.1 V左右的还原电位仍清晰可见。
此外,H-CuF2-SA的倍率性能也远高于H-CuF2-PVDF和CuF2-PVDF(图3e)。图3f比较了迄今为止报道的基于CuF2的转换正极。所有报道的基于CuF2的转换正极在前5个循环中迅速下降,无论在0.2 C高倍率下或在0.02 C极低的电流密度下。与之形成鲜明对比的是,H-CuF2-SA可以实现50个可逆循环,并保持420.4 mAh g-1的容量,在所有基于CuF2的正极中表现出优越的循环稳定性。
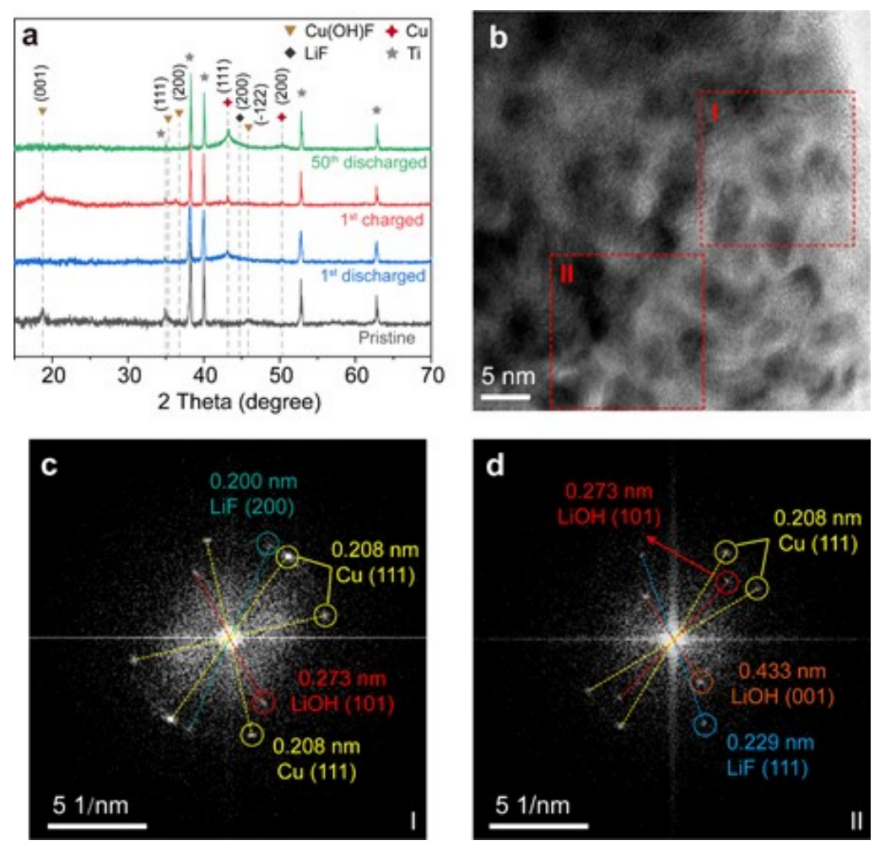
图4 (a)在不同充放电状态下,H-CuF2-SA正极的XRD;(b)第一次放电后,H-CuF2-SA正极的HRTEM图像;(c)对应的区域I和(d)区域II的FFT模式。@ The Authors
作者通过XRD研究了H-CuF2-SA的相变和反应可逆性(图4a)。当H-CuF2-SA完全放电到1.5 V时,与Cu(OH)F的(001)平面相关的18.7°处的峰消失,在43.2°左右出现了一个明显的与Cu的(111)平面对应的宽峰。这表明了Cu(OH)F的完全转化。作者通过HRTEM图像和相应的FFT模式,进一步证明了LiF和LiOH的存在(图4b-d)。由于Cu、LiF和LiOH纳米颗粒的叠加分布,它们在HRTEM图像中的晶格平面存在重叠,难以区分(图4b)。对HRTEM图像的两个选定区域(图4b中的红框)进行了快速傅里叶变换(FFT)分析,以阐明LiF和LiOH的存在。
区域I对应的FFT模式显示了三组衍射点,表明了该区域的多晶分布(图4c)。晶格平面距离为0.208 nm的最亮衍射点与高结晶Cu的(111)平面有关,这与XRD的结果一致。晶格距离较低的暗点与LiF的(200)平面有关。由于(111)平面与Cu的平面距离最大,因此用Cu可以很容易地分辨出表现出较大平面距离的衍射点。因此,在区域I和区域II,位于较小圆上的衍射点可以根据其特征晶格距离分别对应到LiF的(111)平面、LiOH的(101)和(001)平面(图4c,d)。
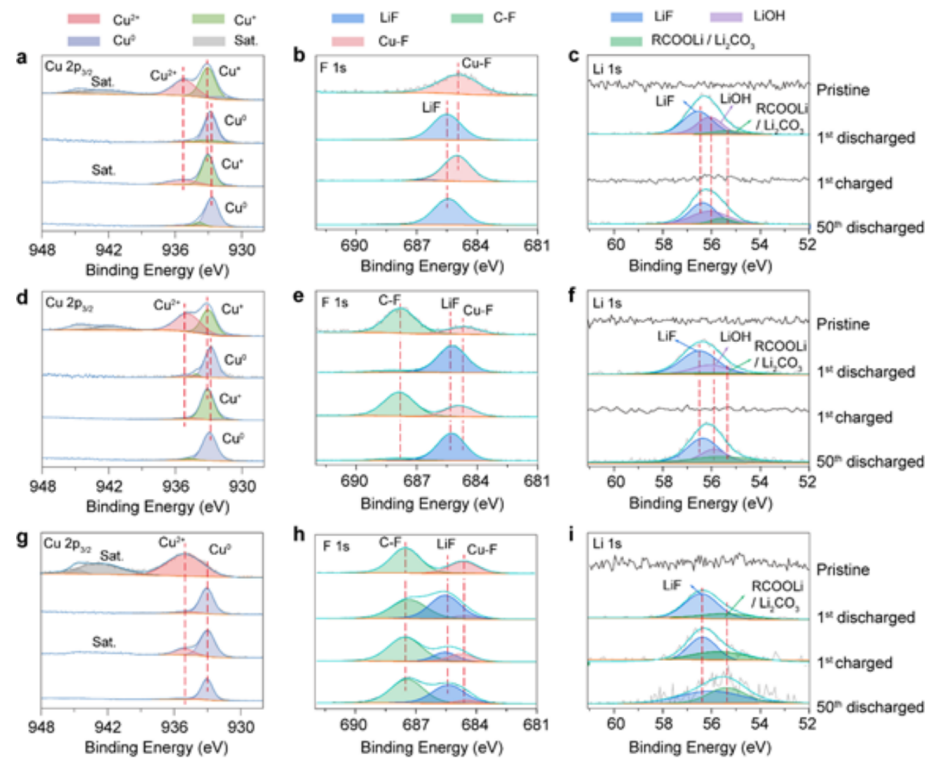
图5(a-c)H-CuF2-SA;(d-f)H-CuF2-PVDF和(g-i)CuF2-PVDF电极循环前后的Cu 2p2/3、F 1s和Li 1s的XPS光谱。每个光谱都从电极表面进行测试。@ The Authors
对原始和循环后的CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA电极进行XPS分析,以揭示它们的可逆性(图5)。对于两个原始的H-CuF2-SA(图5a)和H-CuF2-PVDF(图5d)电极,Cu 2p3/2光谱可以很好地模拟为位于933.1 eV和935.2 eV处的两个峰,这可以分别归因于Cu+和Cu2+的氧化态,表明了铜在Cu(OH)F中的多价性质。而在原始CuF2-PVDF电极中,只观察到一个与Cu2+-F相关的在935.7 eV的单峰(图5g)。
当电池第一次放电至1.5 V,所有三个电极的Cu 2p3/2光谱均移到932.6 eV,对应于金属Cu0,表明这些氟化物电极完全还原。在第一个充满电状态下,原始样品在H-CuF2-SA电极中恢复了Cu2+和Cu+的价态,具有良好的转化反应可逆性。而在H-CuF2-PVDF中,由于电极表面充电过程中铜的严重溶解,Cu2+信号几乎消失,这与其充放电性能相一致。对于CuF2-PVDF电极,Cu0峰的存在表明,由于Cu颗粒的粗化,只有部分金属铜被重新转化为CuF2。纯CuF2的不完全转换主要是由于导电金属铜与分离的LiF聚集体之间的电子传输中断,导致CuF2-PVDF电极中存在大量的“死”活性物质。
放电状态下的Li1s光谱显示了纯CuF2和羟基化CuF2之间不同的转化反应路径(图5c,f,i)。在放电状态下,H-CuF2-PVDF和H-CuF2-SA电极的F1s光谱中都可以检测到Li-F和Li-OH,这有力地证明了在还原反应中形成了LiF和LiOH的混合物,而不是纯LiF。LiOH的存在有效地降低了转化反应的自由焓,从而保证了Cu(OH)F的反应比纯CuF2正极材料更彻底。
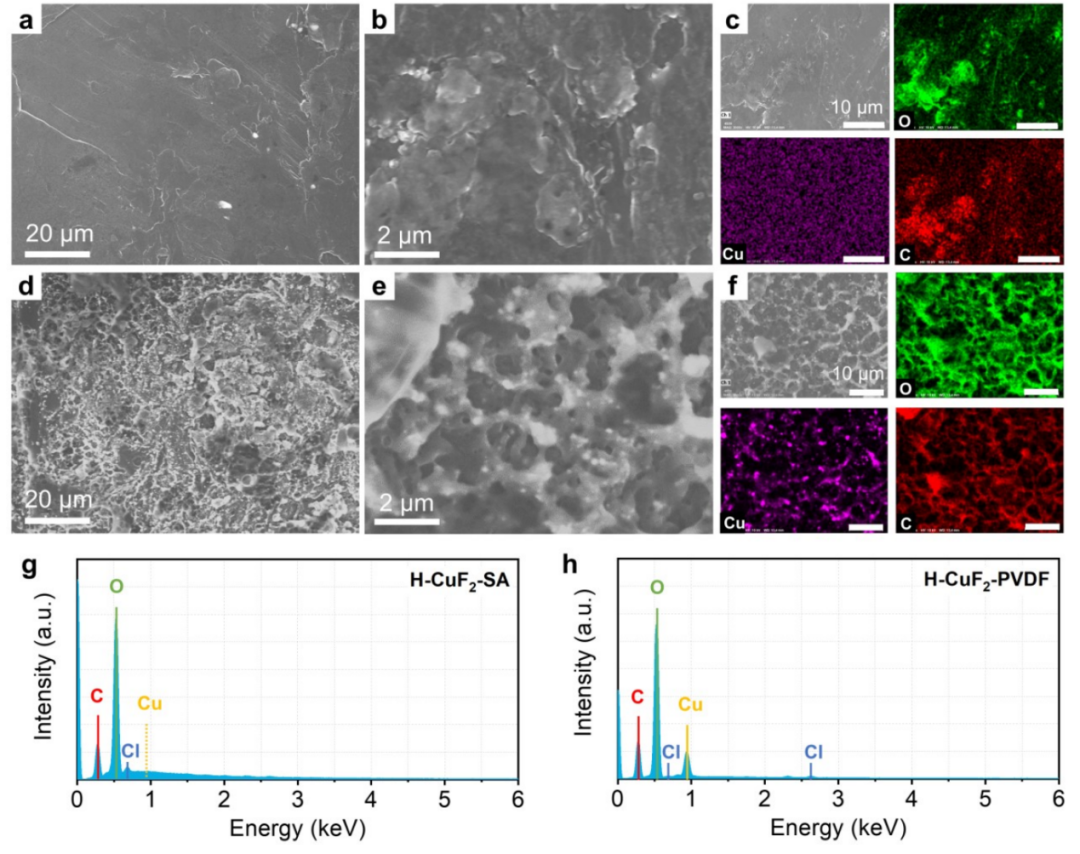
图6 50圈循环后,与(a-c)H-CuF2-SA和(D-F)H-CuF2-PVDF配对的Li负极的SEM图像,及相应的Cu、O、C元素能谱图。(g)H-CuF2-SA和(h)H-CuF2-PVDF配对的Li负极循环后的表面的EDS。@The Authors
由于铜离子在电解液中的溶解会沉积在锂负极上,因此可以用扫描电镜和EDS进行分析。如图6a-b所示,与H-CuF2-SA正极结合的循环后的Li负极,表面光滑,只有少量盐残留可见,图6c所示的C和O元素图像进一步证明了这一点。在相应的EDS光谱中没有观察到Cu信号的痕迹(图6g),这表明H-CuF2-SA的锂化/去锂化过程中没有发生明显的铜溶解。
与之形成鲜明对比的是,与H-CuF2-PVDF正极结合的循环后的Li负极的SEM图像显示,表面极其粗糙,大量的Cu粒子被镀在Li表面(图6d-e)。Cu元素能谱图中的亮蓝色点和相应EDS光谱中的强Cu峰进一步显示了Cu的显著含量(图6f,6h),表明H-CuF2-PVDF中铜溶解严重。
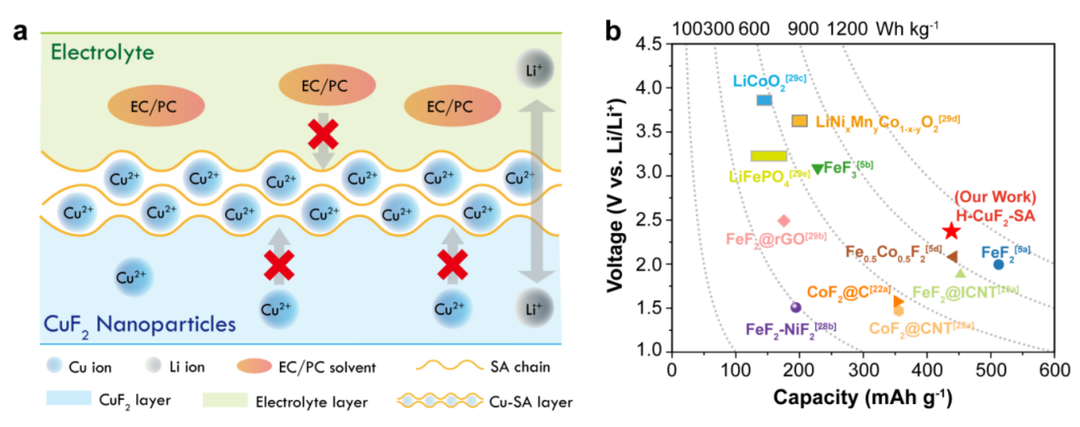
图7 (a)SA粘结剂增强H-CuF2-SA电化学性能的工作机理示意图;(b)H-CuF2-SA与其他金属氟化物和插入正极的容量、电压和能量密度的对比,放电能量密度基于0.05 C的放电电流计算。@The Authors
对H-CuF2-SA的综合表征和电化学分析表明,H-CuF2-SA的超循环稳定性归因于在电极制造过程中原位形成的独特的Cu-SA涂层。图7a示出了H-CuF2颗粒上Cu-SA涂层的形成和功能。在电极制备过程中,由于SA链与水溶液中铜离子之间的强交联效应,在CuF2颗粒表面原位形成了坚固的Cu-SA层。Cu-SA层对Li+有选择性的渗透性,而对Cu2+具有不渗透性,有效地抑制了Cu离子的溶解和降解。此外,Cu-SA层和SA粘合剂在碳酸酯电解质溶剂中的膨胀性都较低,降低了电极/电解质的相互作用,防止了电解液进入粘合剂/电极界面,从而进一步降低了铜在电极中溶解的可能性。
虽然,水基SA粘合剂将水合杂质引入到CuF2材料中,但H-CuF2-SA仍能提供2.4 V以上的平均放电电位。高容量与高放电电压相结合,在50次循环后,电流密度为0.05 C时,可逆放电能量密度为~1009.1 Wh kg-1(图7b),高于大部分金属氟化物。更重要的是,本研究中H-CuF2-SA的能量密度高于大多数铁基氟化物和商用插入正极材料。
05 成果启示
本文通过在CuF2纳米颗粒表面原位形成Cu2+配位SA层,成功地抑制了CuF2正极中Cu离子的溶解。在0.05 C下50次循环后,获得的H-CuF2-SA电极的可逆容量为420.4 mAh g-1,达到1009.1 Wh kg-1的高能量密度。其优越的循环稳定性和能量密度证明了铜基氟化物正极的最佳性能。
审核编辑:刘清
-
LW□-252户外高压交流六氟化硫断路器2024-01-18 4785
-
TE推出SPEC55低氟化物电线和电缆哪里有?-赫联电子2024-03-06 6120
-
六氟化硫断路器常见故障及预防措施2024-12-17 1019
-
三氟化氮的使用2011-05-31 3736
-
六氟化硫断路器有什么优缺点?2019-09-29 3927
-
六氟化硫的性质是什么?2020-03-27 2229
-
详解六氟化硫断路器工作原理2017-11-04 24221
-
六氟化硫传感器的功能特点及应用2021-04-25 2028
-
硅的氧化物和氮化物的气相氟化氢蚀刻作用2022-04-11 2133
-
氟化物分析仪产品手册2022-07-23 554
-
利用海藻酸钠粘结剂和水溶剂制备CuF2电极可以抑制CuF2在有机电解质中的溶解2022-09-07 3683
-
电力系统为何要进行六氟化硫气体在线监测?2023-12-20 1539
-
电力环境六氟化硫气体泄漏监测解决方案2024-06-28 1685
-
电力系统为何要进行六氟化硫气体SF6泄漏在线监测?2024-08-07 1603
-
固态电池革命:Cr-LiF正极材料的循环性能与结构演变解析2026-03-17 792
全部0条评论

快来发表一下你的评论吧 !
