

单原子Cu催化剂的重构现象以及它们在电催化反应的应用
电子说
描述
催化剂的重构现象普遍存在于许多多相反应。目前,借助先进的表征技术以及理论计算,我们已经能够探索、理解多种催化剂的重构机制以及它们对催化反应的利弊。然而,对于单原子催化剂,其重构现象以及机理研究却比较罕见。在许多情况下,我们都默认它们在多相催化反应中是稳定的,特别是在反应初始阶段。
1. 电位驱动单原子Cu催化剂在硝酸盐还原反应的结构演变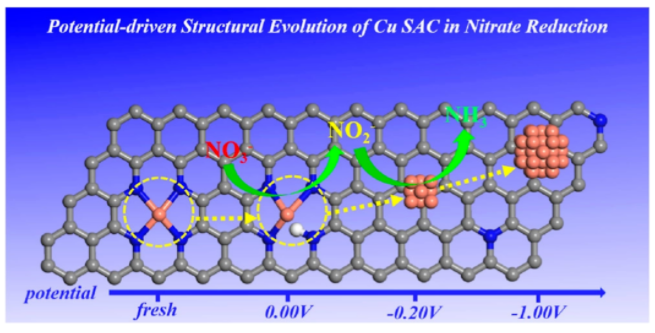
2. 电位驱动单原子Cu催化剂在氧还原反应的结构演变
尽管单原子Cu催化剂的重构现象的研究取得了一定的进展,然而,由于催化反应不同,反应介质以及应用电位也存在差异,目前仍难以清晰解释单原子Cu催化剂的重构现象。
最新成果介绍
德克萨斯大学奥斯汀分校刘远越教授、东南大学王金兰教授等人以单个Cu原子嵌入N掺杂石墨烯为例,利用“恒势混合溶剂化动力学模型”,在实际反应条件下评估了单原子Cu与Cu团簇之间的可逆转化。结果表明,H的吸附是单原子Cu从催化剂表面浸出的重要驱动力。电极电位越负,对H的吸附越强,竞争性析氢反应受到抑制,Cu-N键发生减弱,导致Cu原子部分被锚定在催化剂表面,部分溶解在水溶液中。
在两种状态下Cu原子发生碰撞、形成瞬时Cu团簇结构,成为促进CO2还原为乙醇的真正催化活性位点。当外加电位被除去或转换为正电位时,羟基自由基(OH•)将进一步氧化Cu团簇,Cu通过再沉积、恢复到初始的原子分散状态,最终完成催化剂的重构循环。
因此,该工作提供了对Cu单原子催化剂在工况下的动态稳定性的基本理解,并呼吁考虑现实的反应条件,重新评估目前报道的单原子催化剂的稳定性。
图文介绍
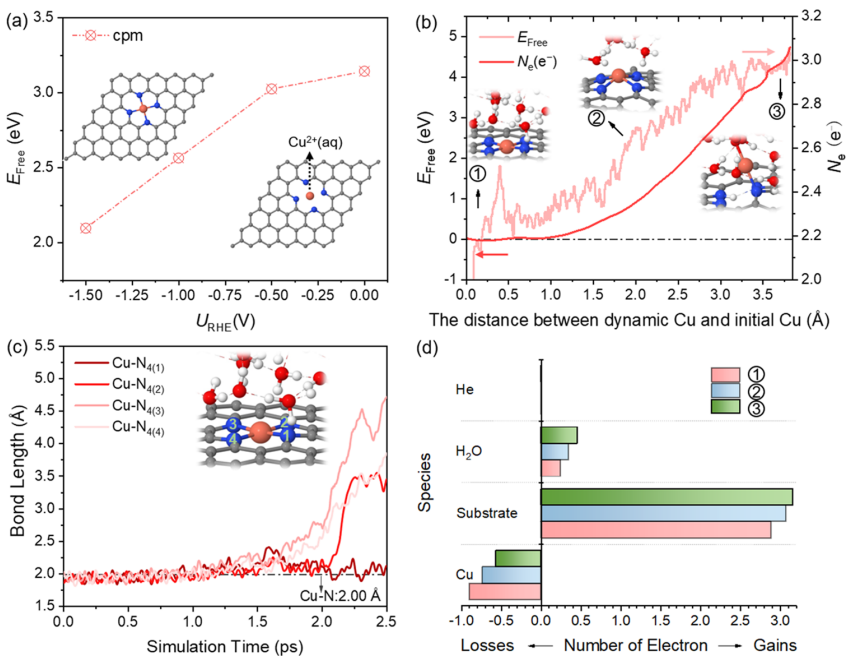
图1. Cu浸出过程的热力学和动力学分析
图1a显示了在0 ~-1.5 V的不同电极电位下,Cu SA从表面浸出、形成Cu2+(aq)的自由能。虽然在-1.5 V时自由能降低到2.09 eV,但从热动力学上仍然很难从表面浸出。进一步通过恒势混合溶剂化动力学模型评估了从表面浸出Cu的动力学可能性。如图1b所示,随着Cu远离表面,自由能继续增加,在反应结束时达到4.74 eV。这表明Cu-N键不易断裂,Cu SA不能在室温下从表面浸出。
图1c跟踪了Cu-N的键长随Cu SA浸出过程的动态演变。注意,当Cu-N(3)键断开时,靠近N(3)原子的一个水分子发生水解,反应结束时对应的构型为Cu原子只与一个N原子和一个−OH基团配位。图1b显示了电子数随结构的变化而变化。与初始结构相比,由于OH -的形成,最终结构的净电荷数增加到约0.7 e-。基于Bader电荷分析,如图1d所示,Cu失去的电子数从初始结构的0.9 e-下降到最终结构的0.56 e-,说明Cu SA发生价态降低、在远离表面处可被还原。因此,热力学和动力学结果都表明Cu SA几乎不可能直接从表面浸出。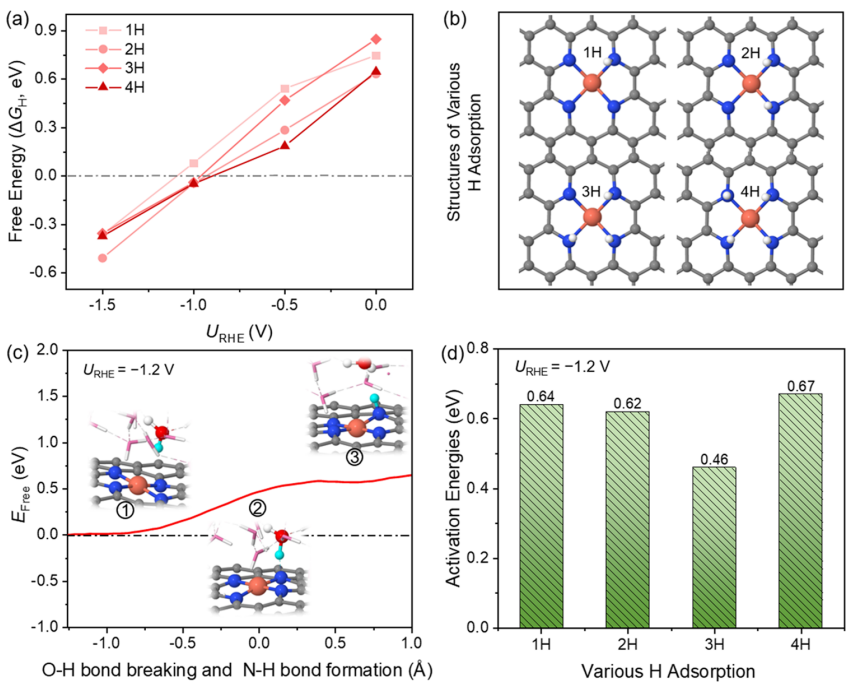
图2. 在催化剂表面的H吸附行为
然而,通过XAS表征,在原位CO2电解条件下原子分散的Cu2+和金属Cu0团簇之间确实存在结构转变。那么,具有强螯合能力的N4-C位点的Cu SA发生浸出、形成Cu团簇的驱动力是什么? 在此,探讨了单原子Cu催化剂在CO2还原电位下H的吸附行为。图2a、b分别显示了不同外加电位下xH吸附的构型与自由能。
当电极电位由零变为负时,对应的H吸附自由能由正变为负,说明H在N位点的吸附从热力学上由不利变为有利。这种转变归因于电位变得更负,导致更多的电子聚集在催化剂表面,从而促进H+的吸附。在U=-1.0 V时ΔGH接近于零,这解释了为什么H2在-1.0 V时产率最高。在U=-1.2 V时,由于H吸附显著增强,析氢反应(HER)被有效抑制。
在此,推测H的吸附可能是影响Cu在负电位下解吸的重要因素。 因此,进一步讨论了H的来源。H的来源与H2O解离有关。因此,计算第一个H2O解离生成H*和OH-的动力学势垒。在−1.2 V时能垒为0.64 eV,说明该反应在室温下很容易发生。反应前后的电子数差约为0.8 e-,证实了OH-的生成。由于催化剂的结构中含有4个N原子,可以为H吸附提供4个活性位点,因此也计算了H占据的其余3个N位点的活化能,分别为0.62、0.46、0.67 eV。H2O解离生成*H和OH-的平均势垒约为0.60 eV。因此,从热力学和动力学两个方面验证了在U=-1.2 V时,H2O分子中的H可以被吸附到N位点上。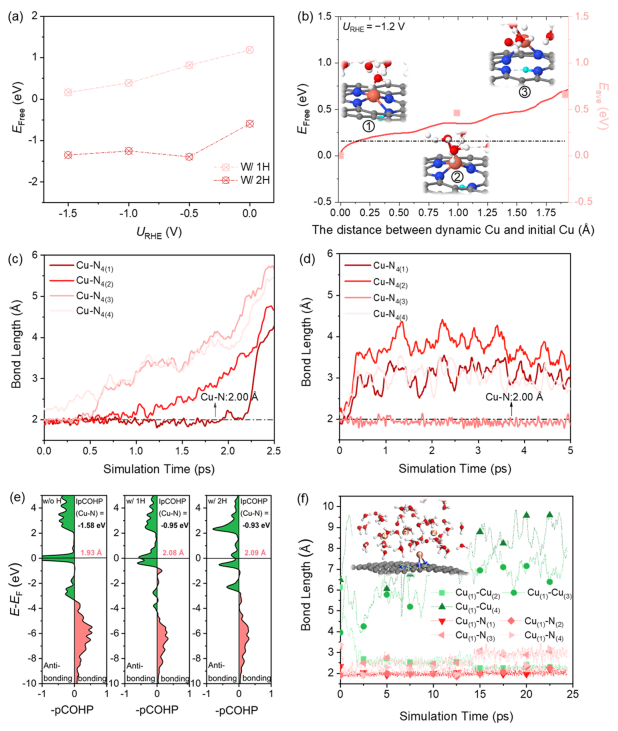
图3. H吸附驱动Cu从催化剂表面发生浸出
接下来,需要考虑的是:在CO2还原过程中,H的吸附如何影响催化剂结构的转变? 计算1H和2H吸附下Cu SA浸出过程中Cu2+(aq)形成的自由能,如图3a所示。随着H的吸附,Cu SA与基底的结合强度减弱,形成了有利于Cu SA浸出的热力学过程。而对于动力学,重新评估了Cu-N4-C中Cu SA浸出过程的动力学势垒,其中一个N位点被一个H位点吸附。如图3b所示,当活化能为0.70 eV时,对应于Cu与一个N原子和一个H2O分子发生配位。与纯Cu-N4-C表面(4.74 eV)相比,H的吸附显著促进Cu SA的浸出。
如图3c所示,当模拟时长为2.25 ps时,Cu原子完全脱离表面,溶解在水溶液中,并吸附两个H2O分子。图3d显示了Cu-N4的键长(x,x=1~4)的动态演变,表明在2H共吸附条件下,Cu SA在短时间内(~ 300 fs)从表面自发浸出。所得到的最终构型由一个Cu-N键和至少一个Cu-O键组成。因此,在动态的电化学界面上,应该同时存在Cu与一个N原子结合的不完全浸出、以及溶解在水中的完全浸出的两种瞬时状态。 在动态环境下,两种瞬时状态的Cu原子发生碰撞形成瞬态Cu3/4团簇结构,成为真正的催化活性中心。进一步模拟了在工作条件下Cu原子的聚集过程。
从图3f中可以观察到,在AIMD过程中,两个Cu原子的聚集小于2.5 ps, Cu(1)原子仍然锚定在N(1)和N(2)上。在15 ps时,Cu(1)-Cu(2)的键长进一步缩短为2.26 Å, Cu(1)-N(3)和Cu(1)-N(4)的键长分别拉伸为3.09和3.03 Å。一旦形成越来越多的Cu小团簇,它们可以加速CO2的还原、形成乙醇。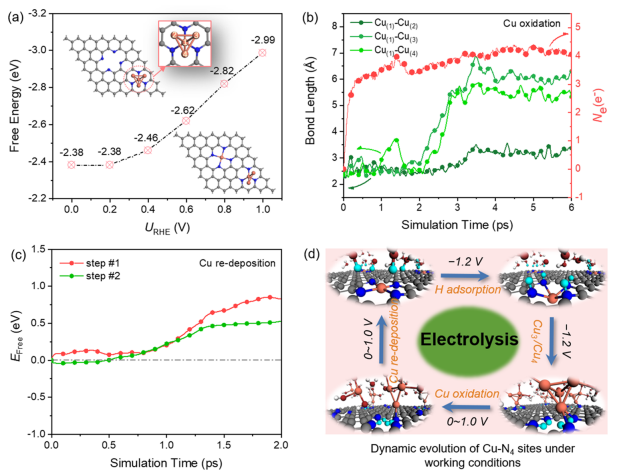
图4. 在工况下Cu-N4位点的动态演化
CO2还原反应结束后,电极电位消失或升高至+1.0 V。此时,Cu团簇可以还原为原子分散的Cu2+。如图4a所示,随着电位的增加,Cu SA-Cu3生成的自由能越负,即Cu SA-Cu3的生成越有利。 最近,一项实验研究证实,HCO3-与H2O之间发生快速氧交换,促进了高氧化性羟基自由基(OH•)的形成,从而促进了Cu的快速再氧化。
因此,考虑在体系中引入两个OH•自由基来评估+1.0 V下Cu4团簇的再氧化过程。如图4b所示,两个OH•自由基分别在2 ps、0.5 ps后氧化Cu-Cu的第一配位壳层。在此之后,Cu(1)原子与其他三个或两个Cu原子的距离越来越远。因此,与纯水溶液相比,OH•自由基的存在在Cu的快速再氧化中起着主导作用。在氧化过程中,系统的净电荷处于相对平衡状态,几乎没有额外的电子转移到电极上,表明Cu团簇被OH•自由基氧化为Cuδ+。
当被OH•氧化后、形成Cu(H2O)3,此时Cu(H2O)3扩散到N4-C位点邻接处时,即第1步,Cu从水溶液中迁移到与N原子配位,动能势垒为0.85 eV,发生了Cu再沉积过程。在这个反应过程中,3个H2O分子返回水溶液中,Cu-N键的平均键长为1.94 Å,与初始Cu-N键长(1.95 Å)一致。也就是说,一旦Cu原子配位到1个N,Cu原子很快就会回到它最初的分散状态。因此,在正电位下,高氧化的OH•与N4-C位点强螯合能力的协同作用,促进Cu团簇恢复到Cu SA状态,完成循环。
审核编辑:刘清
-
燃料电池氧电极催化剂的研究2011-03-11 3119
-
高效电催化二氧化碳还原反应催化剂成功研制2020-03-30 5618
-
高活性生物质碳负载Fe/Pt单原子双功能催化剂开发2021-02-12 3707
-
氧气还原反应催化剂的制作及性能研究2021-08-09 925
-
低结晶和异质结构AuPt-Ru@CNT像高效多功能电催化剂2022-05-31 801
-
调控锂盐消除催化剂表面凝胶化构筑高比能锂硫电池2022-07-13 3065
-
生成CO的催化剂与Cu之间的相互作用2022-08-22 4147
-
Mo配位FeCoNiMo碳负载高熵电催化析氧催化剂图文解析2022-09-20 3588
-
析氢反应(HER)电催化剂在电解装置的广泛应用2022-09-28 12274
-
蜂窝状多孔结晶异质电催化剂实现高效的CO2吸附/活化2022-09-30 4374
-
介绍三金属电催化剂对C2醇的电催化机理2022-10-20 4161
-
应变效应对催化剂活性的影响2022-10-26 3723
-
如何提高HEAs催化剂的催化活性和优选设计研究2022-12-14 1977
-
EnSM:锂硫电池单原子催化剂的基础、应用和机遇2022-12-22 3181
-
双原子催化剂综述:适用于能源和环境催化的双原子催化剂2023-07-17 10710
全部0条评论

快来发表一下你的评论吧 !
