

具有开放孔隙的普鲁士蓝类似物实现高性能锂硫电池
电子说
描述
01 导读
在锂硫(Li-S)电池中,具有开放孔隙的普鲁士蓝类似物(PBAs)通常作为硫的载体,用于缓解充放电过程中的体积变化,抑制多硫化物(LiPS)穿梭,提升电池循环稳定性。另外,最近的一些报道表明,在电池材料中引入高熵(HE)也可以稳定晶体结构,增加其化学和结构多样性,显著提高循环性能。然而,使用HE-PBAs载硫还鲜有报道。
02 成果背景
近日,扬州大学庞欢组在Angew. Chem. Int. Ed.上发表了一篇题为“High-Entropy Prussian Blue Analogues and Their Oxide Family as Sulfur Hosts for Lithium-Sulfur Batteries”的文章,该文章通过一种简单的共沉淀方法系统地合成了一系列PBA(二元PBA到高熵PBA),并通过X射线吸收精细结构光谱研究了PBA中的配位环境。结果证实,所有金属都被成功引入PBA中。电化学实验表明,高熵PBA可锚定LiPS,抑制其穿梭,并可作为催化剂促进多硫化物转化。此外,通过使用PBA作为牺牲前躯体,可以制备从二元到六元的各种纳米立方金属氧化物。
03 关键创新
(1)本工作基于五种金属阳离子(Mn2+、Co2+、Ni2+、Cu2+和Zn2+)和K3[Fe(CN)6],通过简便的共沉淀法成功合成了16种PBA(二元PBA到高熵PBA);
(2)HE-PBA不仅可以锚定多硫化物,抑制多硫化物穿梭效应,而且可以作为催化剂促进多硫化物转化。HE-PBA-S正极在0.1C下循环200次后,容量高达570.9 mAh g-1;
(3)PBA还被用作牺牲前躯体,实现了从二元到六元等多种纳米立方结构金属氧化物的可控合成。
04 核心内容解读
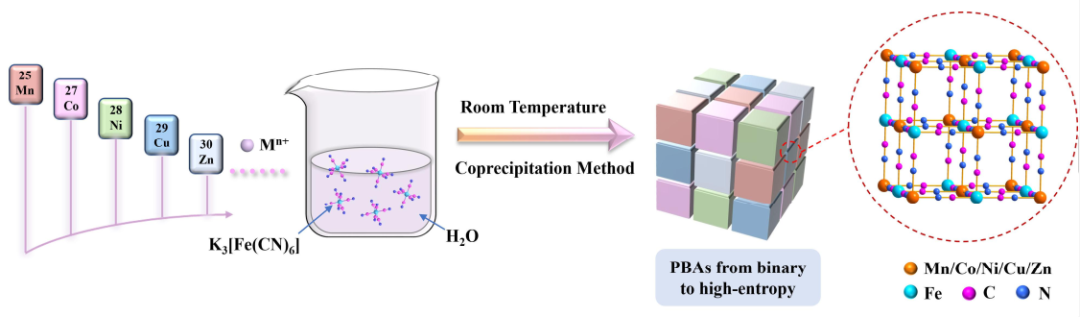
图1. 室温下共沉淀法从二元体系到高熵体系的PBA家族合成过程示意图。@Wiley
图1显示,选择具有五种金属阳离子——MnII、CoII、NiII、CuII和ZnII的硝酸盐作为金属源,以水为溶剂,通过简单的室温共沉淀法,制备了一系列二元、三元、四元和HE PBAs。
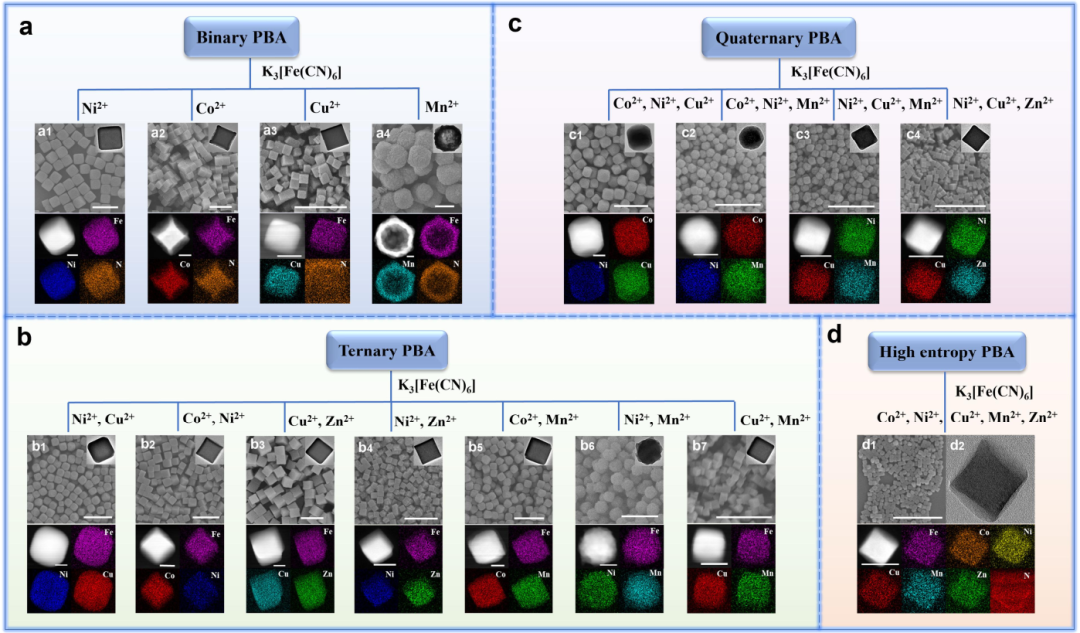
图2. 二元PBA(a1-a4依次代表NiFe-PBA、CoFe-PBA、CuFe-PBA和MnFe-PBA)、三元PBA(b1-b7依次代表NiCuFe-PBA、CoNiFe-PBA、CuZnFe-PBA、NiZnFe-PBA、CoMnFe-PBA、NiMnFe-PBA、CuMnFe-PBA)、四元PBA(c1-c4依次代表CoNiCuFe-PBA、CoNiMnFe-PBA、NiCuMnFe-PBA、和NiCuZnFe-PBA)和HE-PBA(d1和d2代表CoNiCuMnZnFe-PBA)。@Wiley
通过扫描电子显微镜(SEM)和透射电子显微镜(TEM)对形貌进行了表征(图2)。对于二元PBA,NiFe-PBA、CoFe-PBA和CuFe-PBA呈现规则的立方体形状,粒径分别为~310、~300和~120 nm(图2a1-a3)。不同的是,MnFe-PBA呈现出均匀的空心球形状,表面粗糙,粒径约为1 μm(图2a4)。然而,通过这种方法,ZnFe-PBA不会形成沉淀。
对于三元PBA,通过控制Cu/Ni、Ni/Co、Cu/Zn、Zn/Ni、Mn/Co、Mn/Cu和Ni/Mn摩尔比为1:3,成功制备了纳米立方NiCuFe-PBA(~300 nm)、CoNiFe- PBA(~200 nm)、CuZnFe-PBA(~420 nm)、NiZnFe-PBA(~140 nm)、CoMnFe-PBA(~260 nm)、CuMnFe-PBA(~100 nm)和球形NiMnFe-PBA(~ 290 nm)(图2b)。
对于四元PBA,通过控制Co/Ni/Cu、Ni/Cu/Mn、Ni/Cu/Zn和Co/Ni/Mn摩尔比为21分别制备了立方CoNiCuFe-PBA(~330 nm)、NiCuMnFe-PBA(~110 nm)、NiCuZnFe-PBA(~90 nm)和球形CoNiMnFe-PBA(~150 nm)(图2c)。另外,以111的Co/Ni/Cu/Mn/Zn摩尔比,可以制备粒径约为80 nm的均匀立方HE-PBA(图2d)。高角度环形暗场扫描TEM(HAADF-STEM)图像和相应的元素映射分析表明,Mn、Fe、Co、Ni、Cu和Zn在PBA面心立方框架中均匀分布。
综合以上分析发现,合成的球形PBAs均含有Mn源。因此,对于以水为溶剂、无Cu源的共沉淀法合成的PBAs,引入Mn源或增加Mn源含量倾向于合成球形PBAs。
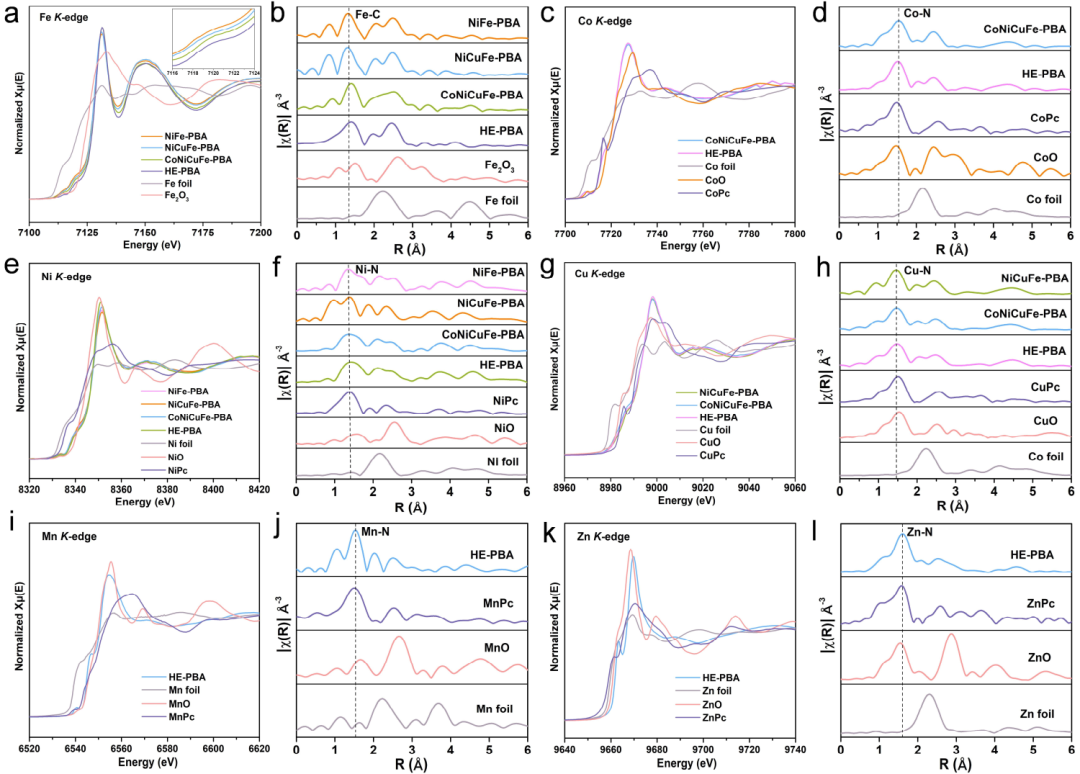
图3. NiFe-PBA、NiCuFe-PBA、CoNiCuFe-PBA和HE-PBA的(a)Fe K边XANES光谱,以及对应的(b)EXAFS光谱。CoNiCuFe-PBA和HE-PBA的(c)Co K边XANES 光谱,以及相应(d)EXAFS光谱。NiFe-PBA、NiCuFe-PBA、CoNiCuFePBA和HE-PBA的(e)Ni K边XANES光谱,以及相应的(f)EXAFS光谱。NiCuFe-PBA、CoNiCuFePBA和HE-PBA的(g)Cu K边XANES光谱,以及相应(h)EXAFS光谱。HE-PBA的(i)Mn K边XANES光谱,以及相应的(j)EXAFS光谱。HE-PBA的(k)Zn K边XANES光谱,以及相应的(l)EXAFS光谱。@Wiley
采用XAFS表征,研究了在PBA骨架中引入Fe、Co、Ni、Cu、Mn和Zn对结构的影响,并判断它们的价态。NiFe-PBA、NiCuFe-PBA、CoNiCuFe-PBA和HE-PBA的Fe K边X射线吸收近边缘结构(XANES)光谱如图3a所示。这些PBA的光谱与Fe箔光谱显著不同,但与Fe2O3光谱相似,表明Fe的氧化态与Fe2O3中的氧化态相似。Fe的吸收边位置按照NiFe-PBA、NiCuFePBA、CoNiCuFe-PBA和HE-PBA的顺序向低能级转移,进一步说明不同金属的掺杂导致了电子性质的变化。不同PBAs中Fe的XANES曲线几乎相同,表明Fe基团所处的配位环境相同。
这些PBAs的Co, Ni, Cu, Mn和Zn K边XANES光谱分别与CoO, NiO, CuO, MnO和ZnO的XANES光谱相似(图3c, e, g, i, k),表明这五种金属元素的稳定价态约为+2。这些PBA的Fe, Co, Ni, Cu, Mn和Zn K边扩展X射线吸收精细结构(EXAFS)光谱中,检测到了明显的Fe-C, Co-N, Ni-N, Cu-N, Mn-N和Zn-N配位(图3b, d, f, h, j和l),进一步表明Fe, Co, Ni, Cu, Mn和Zn成功地引入到PBA骨架中。在这些化学键中,不同金属的引入对Fe-C键长影响较大,而对Co-N、Ni-N和Cu-N键长影响较小。

图4. (a)金属氧化物的合成过程。(b)NiFe-氧化物, (c)CoFe-氧化物, (d)CoNiFe-氧化物, (e)NiCuFe-氧化物, (f)NiZnFe-氧化物, (g)CuZnFe-氧化物, (h)CoMnFe-氧化物,和(i)CoNiCuMnZnFe-氧化物的SEM图像。(j, k)CoNiCuMnZnFe-氧化物的HRTEM图像和SAED图案。(l)CoNiCuMnZnFe-氧化物的HAADF-STEM图像和相应的元素映射。@Wiley
为了进一步拓展PBA材料,本工作还探索了PBA衍生物的可控合成。通过在空气中500 °C下热解NiFe-PBA、CoFe-PBA、CoNiFePBA、NiCuFe-PBA、NiZnFe-PBA、CuZnFe-PBA、CoMnFe-PBA和HE-PBA两小时分别制备了一系列金属氧化物(图4a),包括NiFe-氧化物、CoFe-氧化物、CoNiFe-氧化物、NiCuFe-氧化物、NiZnFe-氧化物、CuZnFe-氧化物、CoMnFe-氧化物和CoNiCuMnZnFe-氧化物。
SEM图像显示,这些氧化物保持其原始立方形貌。TEM图像显示NiCuFe-氧化物、NiZnFe-氧化物和 CoMnFe-氧化物存在中空结构(图4b-i)。高分辨率TEM(HRTEM)图像与选区电子衍射(SAED)图案均证实,CoNiCuMnZnFe-氧化物具有良好的结晶性(图4j, k)。CoNiCuMnZnFe-氧化物的元素映射图像表明,Fe、Co、Ni、Cu、Mn、Zn和O元素均匀分布在多孔纳米立方体中(图4l)。
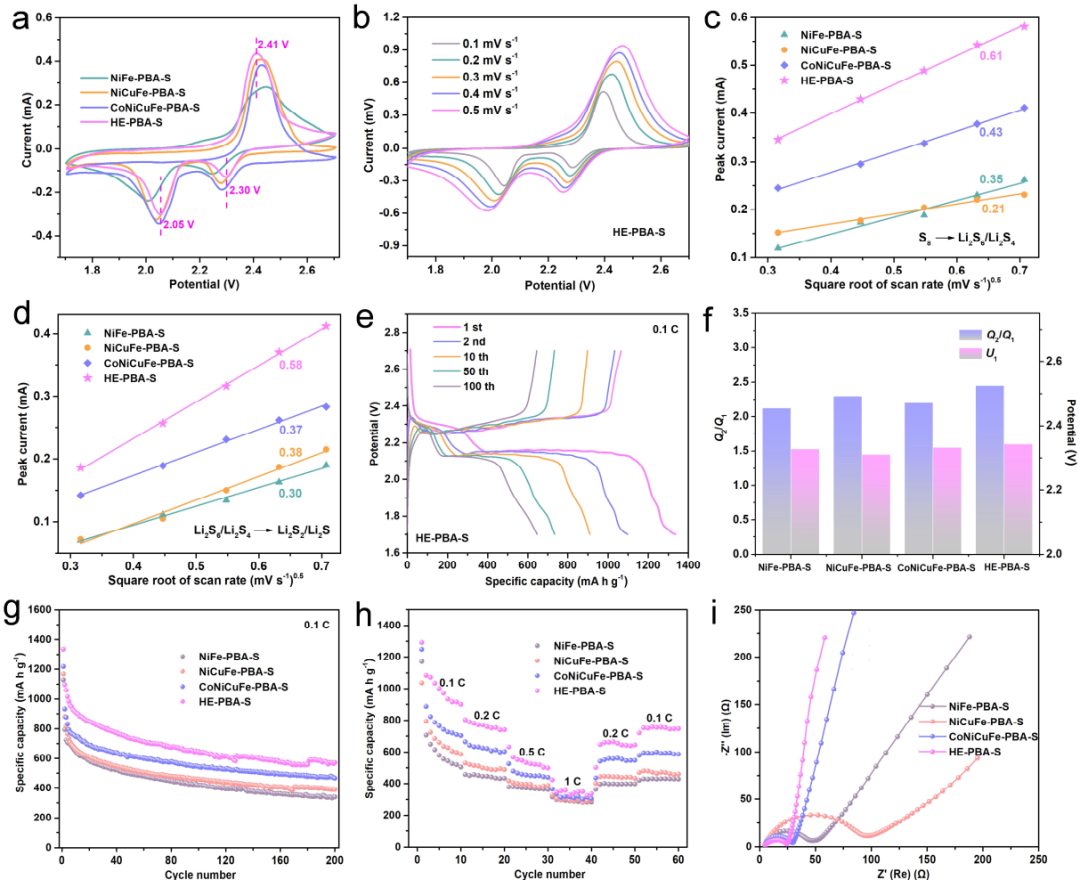
图5. (a)NiFe-PBA-S、NiCuFe-PBA-S、CoNiCuFe-PBA-S和HE-PBA-S正极在0.1 mV s-1扫速下第一次循环的CV曲线。(b)HE-PBA-S正极在不同扫速下的CV曲线。(c)S8转换到Li2S6/Li2S4和(d) Li2S6/Li2S4转换到Li2S2/Li2S的峰电流与扫速平方根(ν0.5)的关系。(e)HE-PBA-S电极在0.1C下的GCD曲线。(f)Q2/Q1和U1的值(从0.1C下的GCD曲线获得)。四个PBA-S样品(g)在0.1C下的循环性能,(h)倍率性能和(i)EIS光谱。@Wiley
通过在130 °C下熔融扩散,硫分子被成功引入NiFe-PBA、NiCuFe-PBA、CoNiCuFe-PBA和HE-PBA的孔中,记为NiFe-PBA-S、NiCuFePBA-S、CoNiCuFe- PBA-S和HE-PBA-S。图5a的CV测试显示,对于HE-PBA-S正极,在2.05和2.30 V处有两个明显的阴极峰,这可归因于从S8分子转变为高阶LiPS并进一步还原为Li2S2和Li2S。
2.41 V的阳极峰可归因于从固体Li2S到LiPS并最终到单质硫的氧化过程。HE-PBA-S正极的CV曲线与其他三个电极相比,还原峰明显正移,氧化峰负移,表明极化程度降低,反应动力学迅速。
图5i的电化学阻抗谱(EIS)显示,NiFePBA-S、NiCuFe-PBA-S、CoNiCuFe-PBA-S和HE-PBA-S正极的奈奎斯特图由高频区域的半圆和低频区域的斜线组成,分别代表电荷转移电阻(Rct)和Warburg阻抗(Wo)。结果显示,HE-PBA-S电极的Rct最小(21.85 Ω),表明电极/电解质界面处的电荷转移增强。HE-PBA-S电极的Warburg斜线更陡,证明其具有更快的Li+扩散过程。
此外,还引入了两个参数U1和Q2/Q1,分别表示放电起始电位和两个电压平台的放电容量之比。在四个PBA-S电极中,HE-PBA-S在0.1C下实现了2.35 V的最高U1和2.45的最高Q2/Q1(图5f),表明HE-PBA载体内的Li+/e-迁移加速。所有这些结果表明,HE-PBA可以促进Li-S电池中硫正极的电荷转移和LiPS氧化还原反应。
为了确认HE-PBA对可溶性多硫化物转化的催化能力,在0.1到0.5 mV s-1的各种扫速下进行了CV测试(图5b)。随着扫速的增加,氧化还原峰的形状没有改变,表明HE-PBA具有良好的电化学稳定性。此外,氧化还原峰电流与扫速的平方根呈线性关系。Li+的扩散系数可以通过Randles-Sevick方程估算:
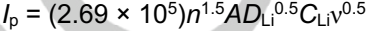
其中Ip代表峰电流,n是电子转移数,A是电极表面积,DLi是Li+扩散系数,CLi是电解质中的Li+浓度,ν是扫速。一般来说,n、A、CLi可以看作是常数。因此,曲线的斜率(Ip/ν0.5)常被用来判断Li+扩散速率。图5c和d显示,无论是S8还原为Li2S6/Li2S4还是Li2S6/Li2S4还原为Li2S2/Li2S,HE-PBA-S电极的斜率均高于NiFePBA-S、NiCuFe-PBA-S和CoNiCuFe-PBA-S电极的斜率,表明HE-PBA-S电极具有更快的Li+扩散速率,且能更有效地实现多硫化物转化。
图5e不同循环下的恒流充放电(GCD)曲线显示,存在两个放电平台,对应S→Li2Sx→Li2S2/Li2S的转变反应。与NiFe-PBA-S(1129.0 mAh g-1)、NiCuFe-PBA-S(1169.1 mAh g-1)和CoNiCuFe-PBA-S(1218.3 mAh g-1)相比,HEPBA-S正极提供了最高的初始放电比容量(1335.6 mAh g-1)。图5g显示,HEPBA-S正极在0.1C下循环200圈后仍然具有570.9 mAh g-1的容量,远优于NiFe-PBA-S(339.5 mAh g-1), NiCuFe-PBA-S(389.8 mAh g-1),和CoNiCuFe-PBA-S(467.0 mAh g-1)。
此外,图5h显示,HEPBA-S表现出良好的倍率性能,其在0.1、0.2、0.5和1C下的可逆容量分别为1292.6、803.5、631.8和424.0 mAh g-1。当电流密度回到0.2和0.1C时,HE-PBA-S电极仍然具有647.4和752.6 mAh g-1的高放电容量,表明在各种倍率下HEPBA-S具有良好的稳定性。
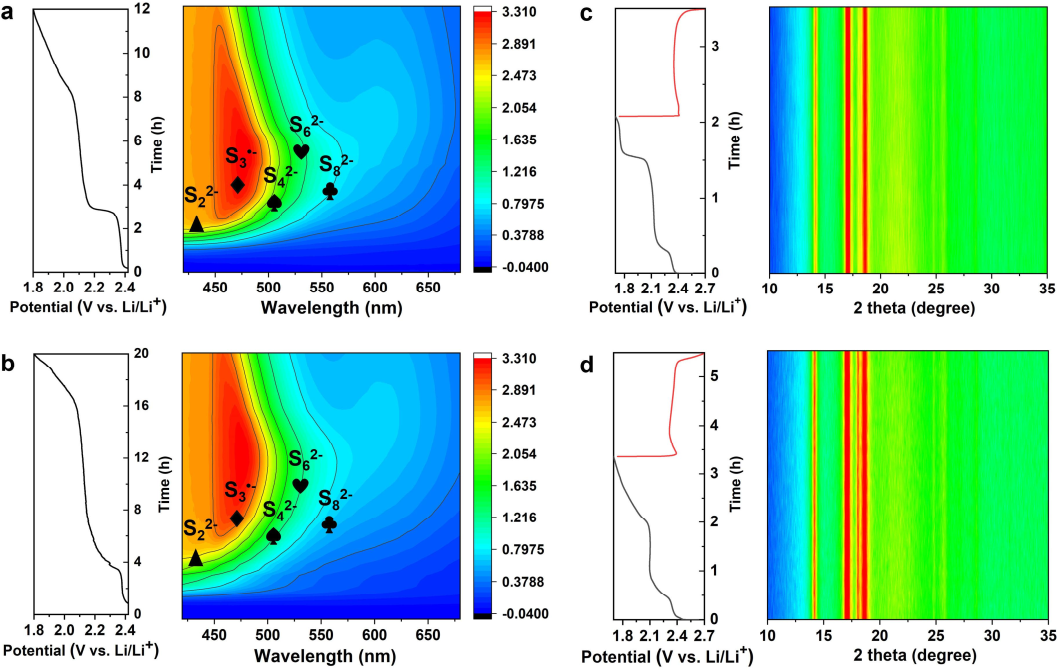
图6. (a)NiFe-PBA-S电极和(b)HE-PBA-S电极的原位UV-vis光谱等高线图和相应的放电曲线。(c)NiFe-PBA-S电极和(d)HE-PBA-S电极的原位XRD等高线图和相应的GCD曲线。@Wiley
图6a和b分别显示了使用NiFe-PBA-S和HE-PBA-S正极材料组装的Li-S电池的紫外可见光谱。在0.05C下第一次放电过程中,出现了各种具有长链或短链的可溶性LiPS。与NiFe-PBA-S电极相比,HE-PBA-S电极具有更长的放电时间。此外,与NiFePBA-S电极相比,HE-PBA-S电极的光谱显示出更高浓度的S62-和S82-,这是由于HE-PBA和LiPS之间的氧化还原反应更快导致。随着放电深度进入第二个电压平台,长链LiPS均呈现一定程度的下降。NiFe-PBA-S和HE-PBA-S在0.1C下第一次GCD过程中的原位XRD图案如图6c和d所示,其中18.6°的峰和22.5°附近的宽峰归因于铝箔。位于14.3°和17.1°的峰归因于PBA晶面。除了这些特征峰外,没有观察到明显的衍射信号,表明含硫物质的含量较低。
05 成果启示
本工作通过一种简便的共沉淀方法在室温下合成了一系列PBA(从二元PBA到高熵PBA)。当用作硫载体时,HE-PBA可以有效锚定多硫化物,并催化其转化来实现高性能Li-S电池。此外,通过使用PBA作为牺牲前体,可以合成各种具有纳米立方结构的金属氧化物。UPS光谱分析表明,CoNiCuMnZnFe-氧化物具有良好的电子特性。这一研究结果为其他PBA和PBA衍生物的合成提供了指导。此外,本研究结果表明,HE材料的构建能够有效提高Li-S电池性能。
审核编辑:刘清
-
新能源汽车电源之锂硫电池利与弊2018-07-13 0
-
锂空气电池未来或击败锂离子电池2018-10-09 0
-
新能源汽车电源之锂硫电池利弊谈2010-11-15 1054
-
锂硫电池的工作原理2017-12-14 43492
-
锂硫电池属于锂离子电池吗_锂离子电池和锂硫电池有什么区别2017-12-14 16887
-
锂硫电池优缺点_锂硫电池电极材料2019-08-23 20991
-
锂硫电池充放电原理_锂硫电池的应用2019-08-23 11343
-
我国锂硫电池技术获新突破2019-09-05 1873
-
UNIST全固态打印双极锂硫电池 容量更大2020-03-24 5023
-
増程发动机是如何打败锂硫固态电池的?2020-12-25 615
-
理论与实验结合建立一种新型的聚合物锂硫电池2020-12-25 570
-
基于锂化普鲁士蓝类似物的锂离子电池正极2022-12-20 1930
-
室温钠硫电池体系硫连续催化转化2022-12-21 900
-
弱化Li+脱溶剂化能垒实现高能低温锂硫电池2023-02-03 1191
-
关于锂硫电池最新研究成果分享2023-12-01 1000
全部0条评论

快来发表一下你的评论吧 !
