

如何从形核机理上破解金属镓的深过冷特性
描述
近日,西北工业大学材料学院凝固技术国家重点实验室先进凝固理论团队及材料基因组国际合作研究中心牛海洋教授与瑞士苏黎世联邦理工学院Michele Parrinello教授团队合作在金属镓的凝固形核机理研究方面取得了新进展,研究成果“金属镓的第一性原理相图及凝固形核研究”(Ab initio phase diagram and nucleation of gallium)近日在Nature Communications在线发表,牛海洋教授和Michele Parrinello教授为共同通讯作者,牛海洋教授为论文的第一作者。
金属镓是一种具有重要工业应用价值的材料,广泛用于半导体、太阳能电池、低熔合金的制造。常压条件下其热力学稳定相α-Ga的熔点只有29.8℃,且其同时具有分子性和金属性。与冰类似,α-Ga的密度低于液态镓的密度。液态镓,尤其是在体积受限条件下,具有极大的过冷度,例如微米尺度的液态镓可过冷至150K的低温。在此种场景下,液态镓凝固结晶得到的是以亚稳相β-Ga为主的混合相而不是α-Ga。如何从形核机理上破解金属镓的深过冷特性以及其凝固过程中α相和β相的竞争之谜已成为困扰该领域内研究人员的科学难题。
从原子尺度出发深入研究形核过程是破解上述难题的关键,然而受限于空间和时间分辨率,目前的实验手段仍难以直接观察材料凝固中晶核的形成过程。随着计算材料科学的不断发展,原子尺度下的计算机模拟,例如分子动力学作为研究复杂凝聚态系统的有力工具,已广泛应用于材料凝固过程的模拟并取得了一系列重大进展。
牛海洋教授与Michele Parrinello教授合作在先进分子动力学方法开发及其在材料凝固研究上的应用方面做出了一系列重要工作(PNAS,2018, 115,5348; JPCL, 2018, 9, 6426;PRL, 2019, 122, 245501)。然而对金属镓的凝固形核过程进行分子动力学计算模拟研究依然需要克服两大困难:其一,液态镓的深过冷特性造成其形核所需的时间远远超过了分子动力学模拟能够达到的时间尺度;其二,受限于金属镓奇异复杂的结构及物理性质,描述金属镓的准确势函数目前依然空缺。
针对上述问题,团队基于材料基因工程的理念,提出将先进分子动力学方法与深度学习方法相结合的研究思路,构建出达到第一性原理精度的金属镓势函数,同时对金属镓的凝固形核过程进行了系统的研究。材料的凝固过程不仅涉及到材料的液固两态,而且还有最核心的中间过渡态,即液-固界面态。研究团队通过采集先进分子动力学方法模拟金属镓的凝固过程中的有效结构信息作为训练集,之后采用第一性原理计算训练集的相关性质,最终通过深度学习算法构建得到计算精度达到第一性原理级别能同时描述金属镓的液态和三种固态结构(α-Ga、β-Ga和Ga-II)的势函数。
采用这一势函数,研究人员确定了包含上述四种结构态的金属镓的温度-压力相图。同时在研究金属镓的凝固机制时发现当温度高于174 K时,β相的形核能垒更低,成功解释了过冷液态金属镓容易形成亚稳态β相而不是稳态α相的实验难题。此外,研究人员将先进分子动力学方法与基于种子技术的分子动力学方法相结合,系统研究了金属镓α相和β相的凝固形核过程并计算了两者的凝固速率。在150 K以上,两相的凝固形核势垒极大且凝固速率极低,与金属镓的深过冷特性相吻合。
上述研究工作不仅推动了人们对金属镓凝固形核机理的深入认识,从理论上破解了金属镓的深过冷特性以及其α相和β相在凝固过程中的竞争规律,并为构建复杂体系的高精度势函数及研究它们的温度-压力相图及凝固形核机理提供了系统性的研究方法。
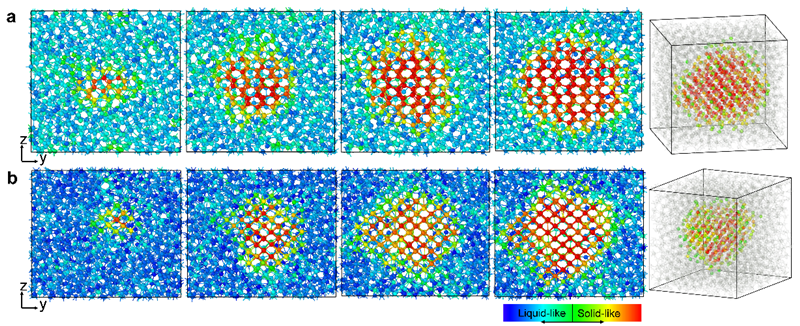
图1 原子尺度下金属镓α相和β相的凝固形核过程
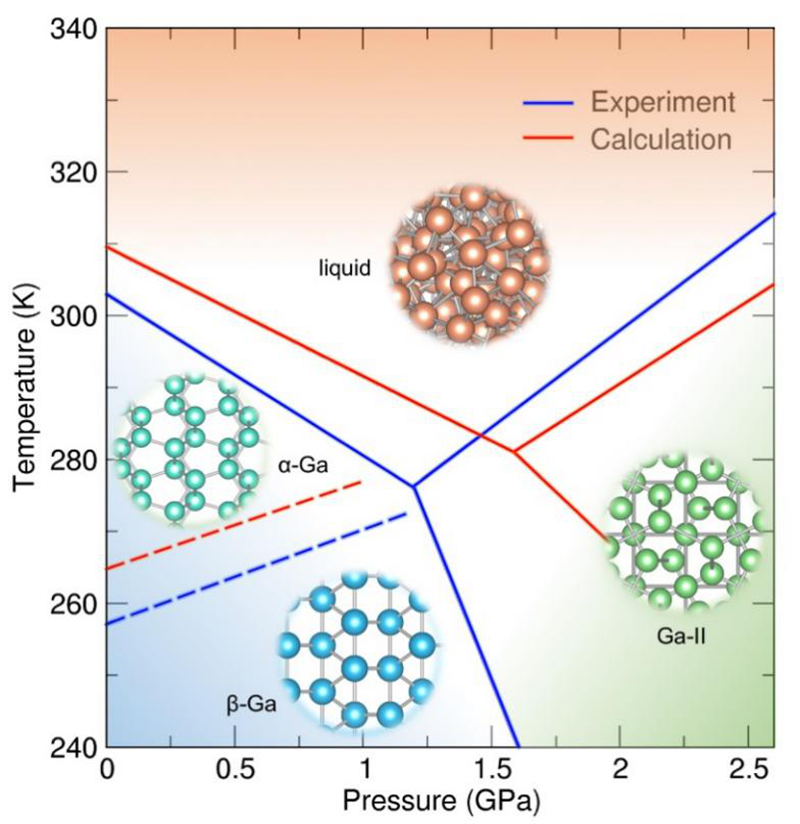
图2 金属镓的温度-压力相图
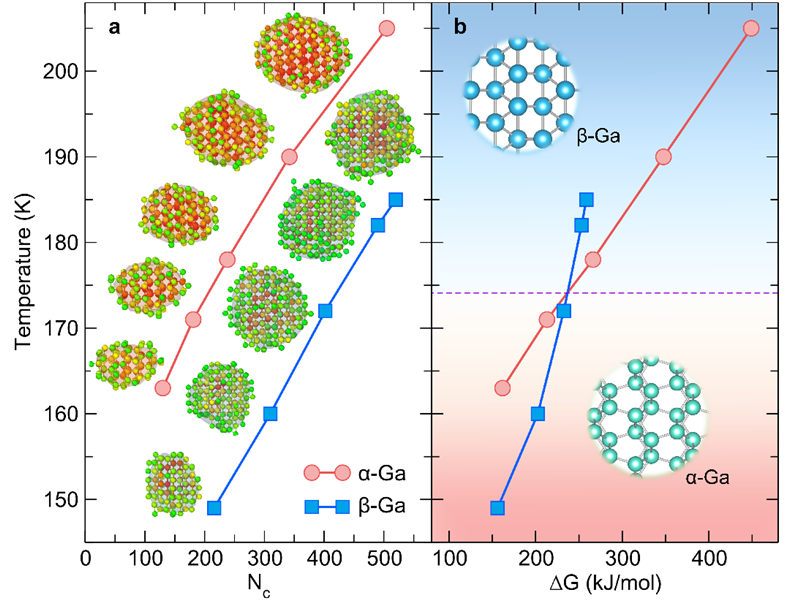
图3 不同温度下金属镓α相和β相的凝固临界晶核及形核势垒曲线图
-
液态金属镓的罐装与包装过程正面临着从传统向智能化转型2024-07-02 842
-
制冷系统如何实现过冷2023-11-08 2012
-
液态金属的驱动特性及其在机器人上的应用2018-10-20 6437
-
中频电源间谐波发射特性机理研究_朱明星2017-01-08 750
-
quartus ip核破解2016-05-19 19065
-
不用加热就能焊接金属?2016-03-10 3992
-
误码特性,误码产生的机理及解决办法2010-03-19 2665
-
海杂波的多重分形关联特性与微弱目标检测2010-02-09 721
-
铜金属锈蚀机理及其电磁线的生产2009-07-03 870
-
金属介电核壳复合粒子的性质2009-03-06 1763
全部0条评论

快来发表一下你的评论吧 !
