

如何解决锌成核/溶解动力学与HER抑制能力之间的内在权衡
电池
描述
01 导读
水系锌离子电池(ZIBs)因其高容量和优异的安全性而有望成为下一代储能技术。然而,锌金属负极在循环过程中会形成枝晶并发生析氢副反应,从而导致电池库仑效率(CE)较低,电极过早失效。对Zn2+溶剂化鞘层进行调控是抑制析氢反应(HER)副反应最直接的方法。但调节溶剂化鞘层会导致Zn2+成核/溶解动力学较差,严重影响Zn电镀/剥离可逆性。因此,如何解决锌成核/溶解动力学与HER抑制能力之间的内在权衡对于开发安全高效的锌离子电池至关重要。
02 成果背景
近日,Angew. Chem. Int. Ed.上发表了一篇题为“Regulating Surface Reaction Kinetics through Ligand Field Effects for Fast and Reversible Aqueous Zinc Batteries”的文章,该文章发现具有中等配体场相互作用的硼酸(BA)可以部分取代Zn2+溶剂化鞘中的H2O分子,形成稳定的缺水溶剂化鞘。它能够实现快速的Zn成核/溶解动力学并显着抑制HER。通过系统地比较BA和其他电解质添加剂的配体场强和溶剂化能,发现溶剂化能与Zn成核/溶解动力学和HER抑制能力有很强的相关性。该工作为锌电池溶剂化鞘的设计提供了参考。
03 关键创新
(1)BA可以部分取代Zn2+第一配位层中的水分子,形成新的缺水溶剂化鞘。
(2)原位光学显微镜和有限元分析(FEA)表明,BA还可以调节Zn电极表面周围的电场,以实现均匀的Zn沉积。
(3)具有BA添加剂的Zn//Zn对称电池可以在0.5 mA cm-2下稳定循环超过3000小时。即使在5 mA cm-2下,它也能提供84 mV的超低电压极化和5.25 Ah cm-2的累积电镀容量(CPC),表明Zn2+成核/溶解动力学显着改善。
04 核心内容解读
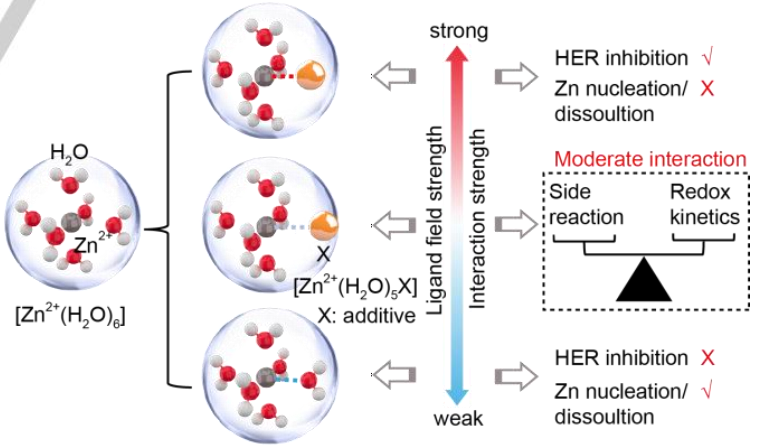
图1. Zn2+和添加剂之间的配体场强、相应的溶剂化鞘层以及它们对HER和Zn成核/溶解反应动力学的影响。@Wiley
基于经典配体场理论和Sabatier原理,寻找合适的配体,进而调节配体场强度和相应的氧化还原动力学在理论上是可行的。配体场强与副反应动力学的关系如图1所示。其中配位作用过弱的配体不能稳定缺水的溶剂化鞘,导致有害的HER副反应,而过于稳定的溶剂化鞘则导致解溶剂化过程受阻,成核/溶解动力学迟缓。
因此,需要寻找具有适度配体场效应的新型电解质添加剂来平衡副反应和氧化还原动力学之间的矛盾。硼酸(BA)具有比大多数有机小分子更宽的氧化电位和略弱的配位能力,由于形成反馈π键,预计其与锌离子具有适度的配体场相互作用。
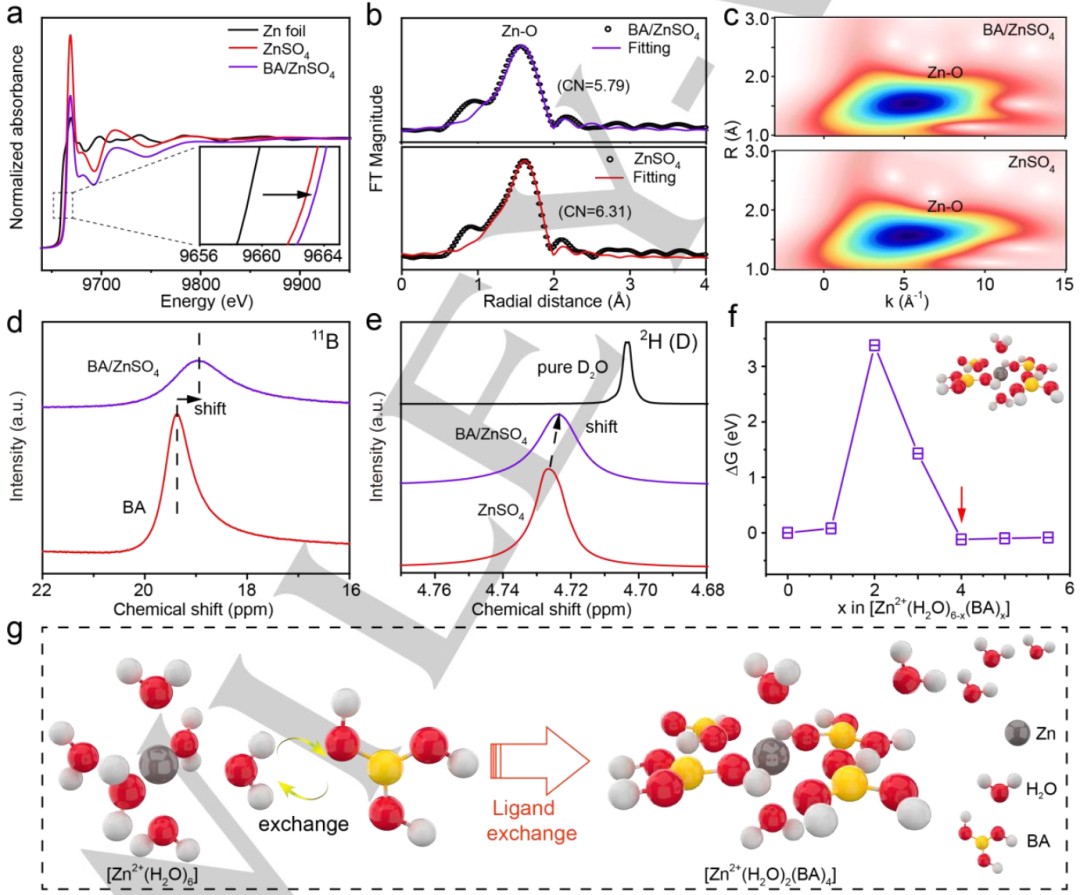
图2. a)归一化的Zn K边XANES谱。插图:放大的Zn K边XANES谱。b)R空间中的EXAFS谱。c)EXAFS谱的小波变换图像。d)BA/ZnSO4电解液和BA溶液的11B NMR谱。e)BA/ZnSO4电解液、ZnSO4电解液和纯D2O中的2H NMR谱。f)所有可能的Zn2+溶剂化鞘的ΔG。插图是稳定的[Zn2+(H2O)2(BA)4]溶剂化鞘。g)引入BA后Zn2+溶剂化鞘的配体交换过程示意图。@Wiley
为了揭示BA添加剂对Zn2+溶剂化鞘层的影响,进行了XAFS、拉曼光谱和 NMR测试。图2a显示了BA/ZnSO4电解质、Zn箔和ZnSO4电解质的Zn K边X射线吸收近边结构(XANES)谱。由于其固有的金属特性,锌箔具有最小的吸收边能量。ZnSO4放大的Zn K边XANES曲线显示,在引入BA后,近边吸收能量转移到高能区,表明BA/ZnSO4电解质中Zn2+周围的平均电子密度低于ZnSO4电解液。
此外,利用Zn K边XANES谱在R空间的傅里叶变换(FT-EXAFS)探究BA/ZnSO4电解质和ZnSO4电解质的局部溶剂化鞘(图2b)。~1.60 Å处的单个第一配位壳来源于Zn-O矢量。BA/ZnSO4电解液和ZnSO4电解液中Zn-O径向距离分别为2.05和2.08 Å,Zn-O的配位数(CN)分别为5.79和6.31。此外,小波变换(WT)EXAFS用于分析Zn-O配位(图2c)。CN和WT最大值相似,说明引入BA后仍为Zn-O配位。
此外,进行核磁共振谱以评估Zn2+和BA分子之间的相互作用。图2d显示,BA水溶液的11B峰位于19.372 ppm。添加2.0 M ZnSO4后,11B峰移至18.933 ppm,表明BA分子中B原子周围电子密度增加,屏蔽效应增强。这种现象可以归因于BA分子在Zn2+溶剂化鞘层中的取代。
此外,在核磁共振谱中,纯D2O的2H峰通常位于4.703 ppm(图2e)。引入2.0 M ZnSO4后,2H峰升高,达到4.727 ppm,说明2H的电子密度增加是由于Zn2+和D2O的配位作用导致。如果进一步向体系中加入BA分子,水的2H峰开始向前移动到4.723 ppm,表明一些被束缚的水分子再次被释放。
为了进一步探究溶剂化鞘结构,进行了理论计算。考虑了各种BA和H2O比例下所有可能的溶剂化鞘结构,并计算了相应的吉布斯自由能(ΔG),如2f所示。根据计算结果,BA由于ΔG最低,容易取代4个水分子形成新的缺水溶剂化鞘,表明配体交换在热力学上是有利的。这些结果说明BA添加剂通过配体交换改变了溶剂化鞘,如图2g所示。
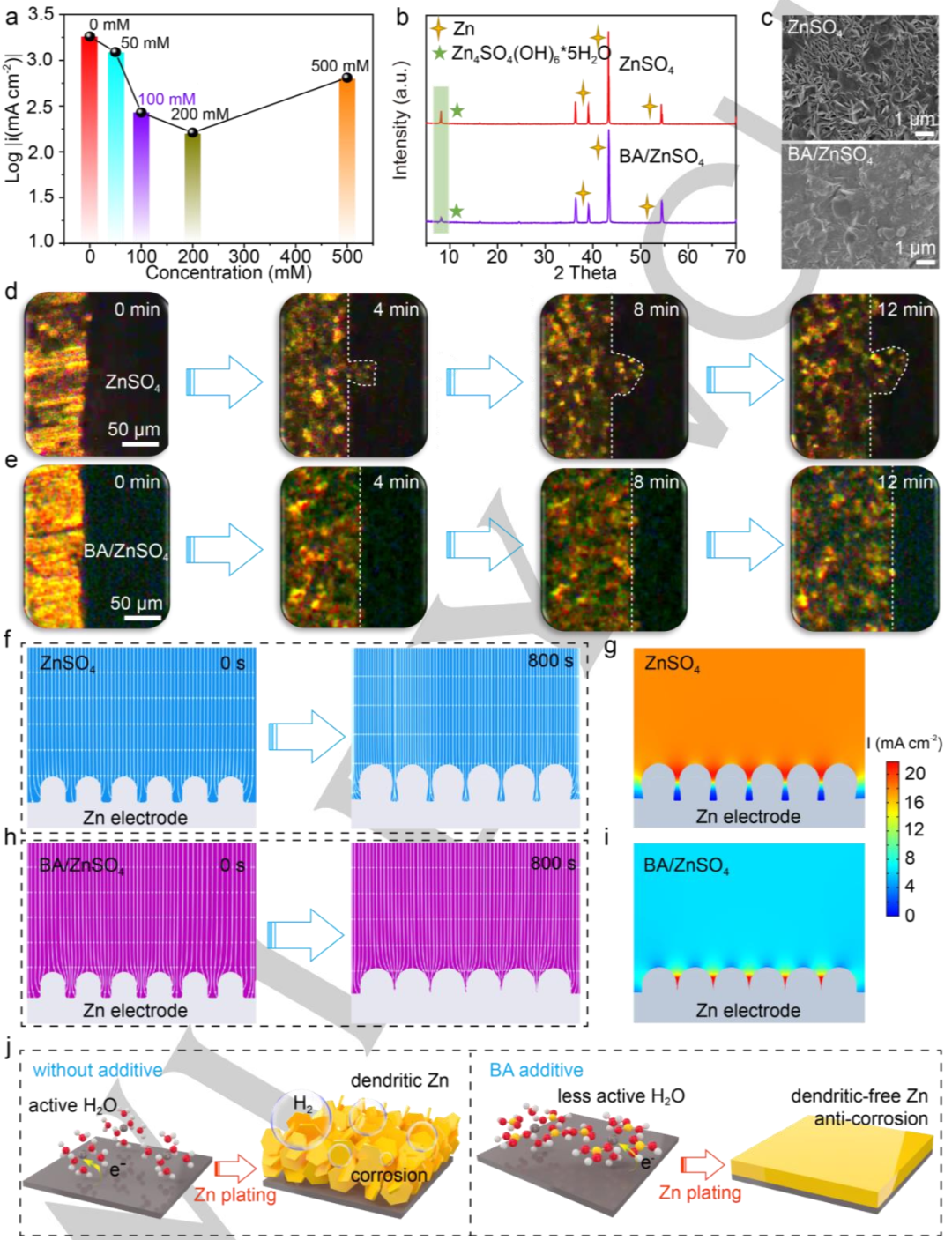
图3. a)不同溶液下的腐蚀电流密度。具有ZnSO4和BA/ZnSO4电解质的Zn//Cu电池循环50次后,Zn电极的b)XRD谱和c)SEM图。d)ZnSO4,e)BA/ZnSO4电解质中,锌沉积行为的原位光学显微镜研究。f,g)ZnSO4电解质,h,i)BA/ZnSO4电解质中,电极/电解质界面的FEA和相应的电流密度分布。j)BA添加剂在ZnSO4电解质中对Zn负极界面反应的改性机理示意图。@Wiley
图3a显示,含有BA添加剂的电解质表现出较低的腐蚀电流密度,表明锌负极的耐腐蚀性能得到增强。此外,利用X射线衍射(XRD)对在Zn//Cu电池中经过50次电镀/剥离循环后的Zn电极进行表征(图3b)。在8.1°附近的峰,与副产物Zn4SO4(OH)6·5H2O相关,在添加BA后,该峰强度显着降低,再次证实了BA对腐蚀的抑制作用。
图3c的扫描电子显微镜(SEM)图像显示,由于锌与水系电解质之间的连续反应,没有BA添加剂的锌循环后,表面变得粗糙,存在片状枝晶。相比之下,在BA/ZnSO4电解液中循环的Zn表现出致密和光滑的形貌。因此,BA添加有望调节锌沉积行为。
为了进一步研究溶剂化鞘对锌沉积行为的影响,采用原位光学显微镜直接观察电解质-电极界面处的锌沉积形貌。对于没有BA添加剂的Zn电极,在40 mA cm-2下沉积4分钟后会出现不均匀的成核位点(图3d)。这些突出的锌位点不受控制地长成枝晶,最终导致循环可逆性差和电池失效。相反,使用BA添加剂,沉积过程看起来均匀且稳定(图3e)。电镀12分钟后,没有观察到类似枝晶的形貌,表明添加BA有利于使成核位点均匀化,最终抑制了Zn枝晶的生长。
有限元分析(FEA)用于进一步了解了BA添加剂对Zn沉积行为的影响。图3f显示,在没有BA添加剂的情况下,Zn的连续沉积往往会发生在不平整的表面上,并最终导致无法控制的生长。此外,枝晶尖端的电流强度明显高于其他区域,表明枝晶将进一步生长(图3g)。相比之下,锌尖端的电场和电流密度被BA添加剂屏蔽,使锌能够均匀沉积(图3h和3i)。基于上述结果,BA在增强耐腐蚀性和抑制HER副反应中的作用如图3j所示。
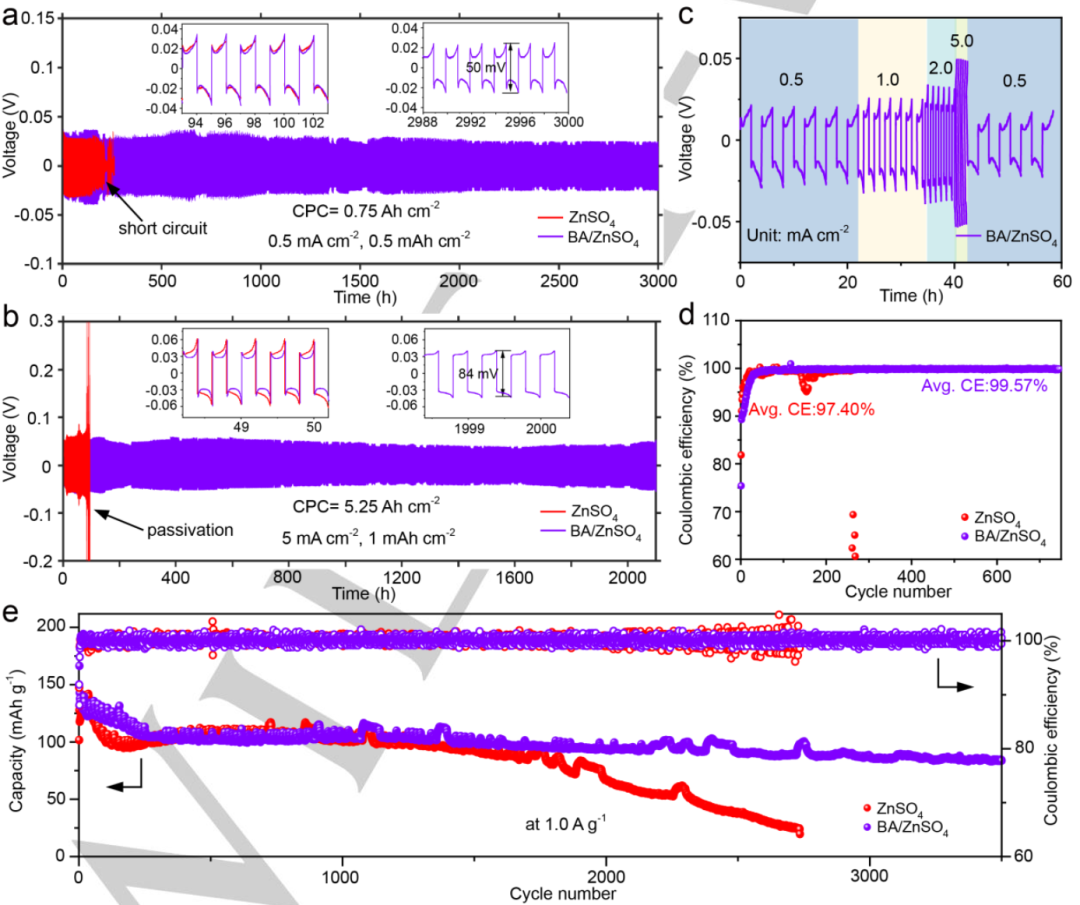
图4.在a)0.5 mA cm-2、0.5 mAh cm-2和b)5 mA cm-2、1 mAh cm-2下添加或不添加BA的Zn//Zn对称电池循环性能。c)使用BA/ZnSO4电解质的Zn//Zn对称电池倍率性能。d)不同电解液中,1 mA cm-2@0.5 mAh cm-2下Zn电镀/剥离的CE。e)具有两种电解质的Zn//MnO2电池在1.0 A g-1下的循环稳定性和效率。@Wiley
图4a显示,添加BA添加剂后,Zn//Zn对称电池在0.5 mA cm-2@0.5 mAh cm-2条件下可以稳定循环超过3000小时,而没有BA添加剂的电池在210小时后会短路。即使在更高的电流密度(5 mA cm-2@1 mAh cm-2)(1.9% 的放电深度,DOD)下循环,使用BA/ZnSO4电解质的对称电池也能实现2100小时的超长循环寿命(图4b),几乎是没有BA添加剂(约100小时)的21倍。图4c显示,使用BA添加剂后,Zn//Zn对称电池在所有电流密度下都表现出稳定的电压曲线。
图4d显示,在没有BA添加剂的Zn//Cu电池前268个循环中,平均CE为97.4%。但CE值在随后的循环中波动。相比之下,具有BA添加剂的Zn//Cu电池CE明显提高,平均值为99.57%,这是因为BA取代了溶剂化鞘中活性较低的水分子。图4e显示,添加BA添加剂的电池可以稳定循环3500次,进一步证明BA可以显着提高锌电池的循环稳定性。
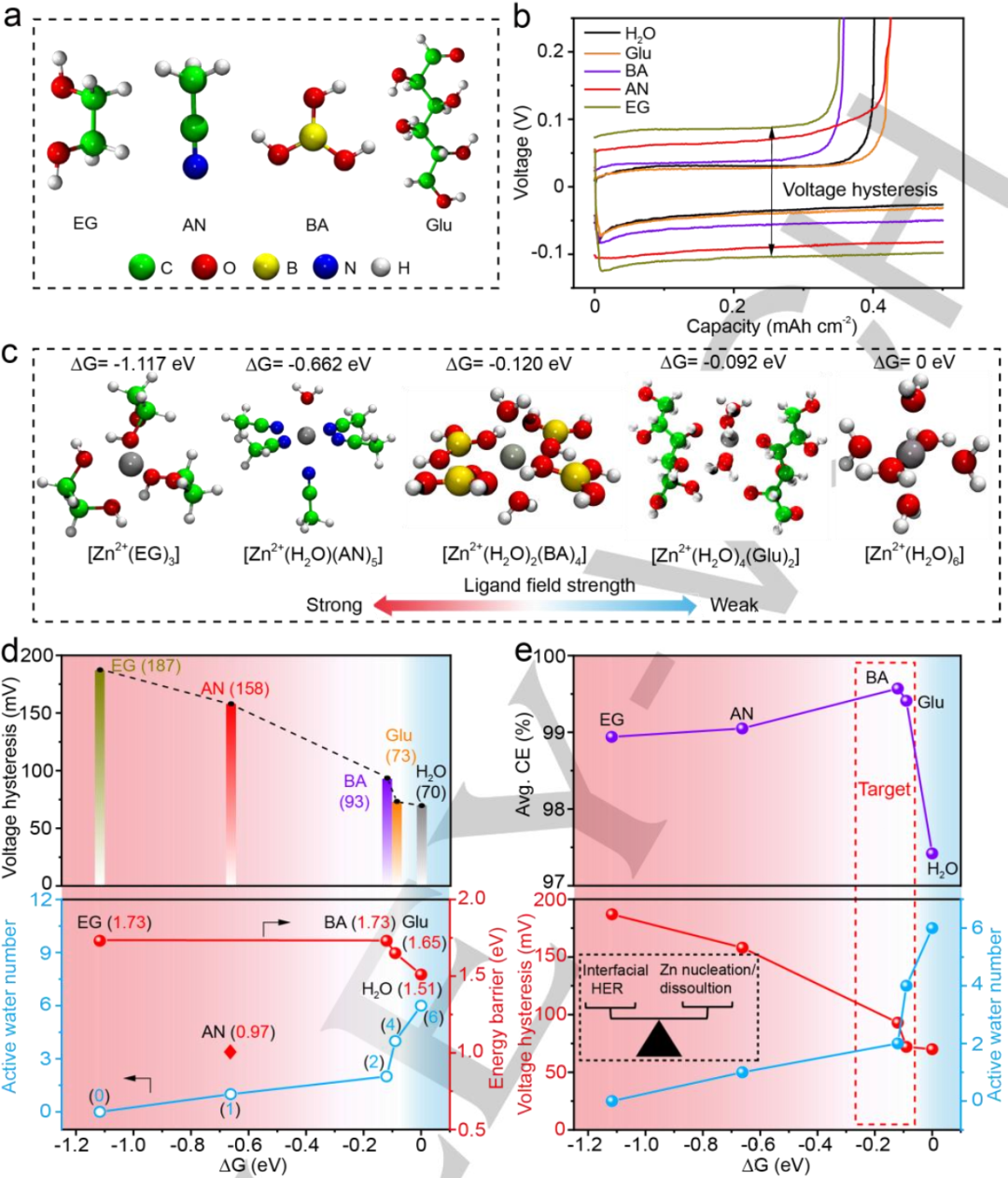
图5. a)不同添加剂的结构。b)引入不同添加剂后第一个循环的电压曲线。c)不同添加剂存在下优化的Zn2+溶剂化鞘和相应的配体场强顺序。d)溶剂化鞘ΔG、Zn成核/溶解动力学(顶部)和界面HER(底部)之间的相关性。e)ΔG、电压极化、溶剂化鞘中的活性水数量和平均CE之间的相关性。@Wiley
接下来,利用DFT计算系统地评估了溶剂化鞘的热力学稳定性和水解离动力学。除了BA分子外,还考虑了文献中常用的添加剂EG、AN、和Glu分子,如图5a所示。图5b显示了添加了EG、AN、BA、Glu和H2O的Zn//Cu半电池电压极化。其中EG、AN、BA、Glu和H2O的电压极化分别为187、158、93、73和70 mV,说明添加剂的引入显著提高了Zn成核/溶解的能垒。
为了进一步探究各种添加剂与Zn2+在Zn成核/溶解过程中的相互作用,计算了溶剂化结构的吉布斯自由能(ΔG)。在不同添加剂作用下,最稳定的溶剂化结构如图5c所示,其中EG、AN和Glu的电解质添加剂更倾向于与Zn2+配位,分别形成稳定的[Zn2+(EG)3]、[Zn2+(H2O)(AN)5]和[Zn2+(H2O)4(Glu)2]溶剂化鞘。溶剂化鞘对应的ΔG值顺序为EG>AN>BA>Glu>H2O。
此外,Zn2+核与周围配体分子之间的溶剂化鞘基本可以用配体场理论来描述。具有双齿配体的EG表现出最强的配体场,而具有弱ơ键的水表现出最弱的配体场。其中BA配体由于π*反馈键的形成,具有适度的配体场。因此,形成的稳定溶剂化鞘配体场强度顺序与ΔG的趋势很好地吻合。
为了将计算得到的溶剂化鞘ΔG与Zn成核/溶解动力学和HER抑制能力联系起来,图5d绘制了溶剂化鞘的活性水数量、电压极化与ΔG之间的关系。ΔG与[Zn2+(EG)3],[Zn2+(H2O)(AN)5],[Zn2+(H2O)2(BA)4],[Zn2+(H2O)4(Glu)2]和[Zn2+(H2O)6]的溶剂化鞘初始电压极化呈正相关。
此外,随着配体场的增大,ΔG减小,相应的电压极化变大。其中[Zn2+(H2O)6]由于初始极化电压较小,具有最佳的Zn2+成核溶解动力学。但是,第一溶剂化壳层中水分子的充分占据会大大加速Zn表面的HER动力学。因此,接下来探讨了电解质添加剂对界面HER动力学的影响。如图5d(下)所示,较强的配体场对应较低的ΔG,而在原始的[Zn2+(H2O)6]溶剂化鞘中,更多的水分子被配体添加剂取代。
图5e显示,ΔG不仅与初始电压极化具有很强的相关性,而且与活性水数量和平均CE呈火山状关系,这意味着理想的溶剂化鞘不应具有太强或太弱的 ΔG(以及配体场强),从而实现出色的Zn成核/溶解动力学并抑制HER副反应。
05 成果启示
该工作证明了BA配体与Zn2+核具有适度的配体场相互作用,从而实现了良好的Zn成核/溶解动力学并抑制了水的解离。XAFS和NMR测试表明,BA仅通过配体交换部分取代Zn2+配位层中的水分子,形成了一个稳定的缺水溶剂化鞘。添加BA的Zn//Zn对称电池在5 mA cm-2下能够实现5.25 Ah cm-2的累积电镀容量和84 mV的超低电压极化。
BA和之前报道的电解质添加剂BA、AN、EG、Glu的配体场强度相比,Zn2+溶剂化鞘的相对自由能与Zn成核/溶解动力学和HER抑制能力有很强的相关性,表现出典型的火山行为。配体场强与副反应动力学之间的相关性为合理设计快速和可逆的锌电池提供了新的思路。
审核编辑:刘清
-
Adams多体动力学仿真解决方案全面解析2025-04-17 7773
-
轮毂电机驱动电动汽车垂向动力学控制研究综述2025-03-07 418
-
刚性机械臂的动力学建模2023-11-17 1697
-
基于车辆动力学模型的横向控制2023-11-15 1836
-
机械动力学原理.pdf2021-11-04 1723
-
分布式驱动电动汽车的动力学控制有哪几种类型?常见问题是什么?2021-08-30 1296
-
飞行器动力学参数在线辨识EKF算法实验流程2021-08-27 1368
-
电动力学或经典电动力学统称为什么?2020-06-10 4549
-
汽车系统动力学长篇大论2017-11-01 4982
-
飞盘空气动力学2016-12-25 1631
-
基于多体系统动力学的空气悬架大客车平顺性试验仿真研究2009-12-02 4065
-
热分析动力学2009-12-01 681
-
[下载]想了解多体动力学软件吗?有教程分享及免费试用下载2009-03-24 4226
-
电力拖动系统的动力学课件2008-11-19 4382
全部0条评论

快来发表一下你的评论吧 !
