

一种新的xLi3P−(1−x)Li2S固溶体的合成方法
电池
描述
【研究背景】
锂离子电池(LIBs)的能量密度和电压较高,是最有前途的存储系统之一。对于LIBs综合性能的进一步提升,锂金属负极的使用是必然趋势。然而,锂金属化学活性较高,在传统的有机电解液体系中更容易引发安全风险。因此,全固态电池(ASSBs)应运而生。然而,目前缺少商业化的高导电性固态电解质,同时无枝晶的锂金属可逆循环也是一大问题。
关于高性能固态电解质的研发目前已有一定进展,如阳离子取代的Li7La3Zr2O (LLZO)衍生化合物,离子电导率高,对锂金属的动力学稳定性,可以在空气气氛中工作。然而,其容易开裂,机械性能较差,对锂枝晶的形成无法形成弹性缓释。硫化物虽然对水敏感且不耐氧化,然而其离子电导率较高,且机械性能好。富锂三元磷化物目前也被认为是有前景的固态电解质,P3-比硫化物阴离子具有更大的极化率。
尽管上述固态电解质各有千秋,却有一个共性问题—对金属锂本征热力学不稳定。因此,开发兼具高离子电导和对金属锂稳定性高的固态电解质,必须综合考虑结构和热力学特性以及合成方法。
【成果简介】
作者利用高通量晶体结构预测辅助简单的高能球磨技术,将Li3P和Li2S全无序化以制备固态电解质,并通过多种表征分析方法证明其不仅具备高离子电导率,对于金属锂还具备本征的热力学稳定性。该工作为高性能固态电解质的开发提供了理论指导和方法借鉴。
【研究亮点】
(1)利用高通量晶体结构预测技术,首次预测并发现Li-P-S三元体系具备高性能固态电解质的潜质,并绘制出相图。
(2)利用简单的固相球磨法使Li3P和Li2S完全无序化,制得的固态电解质具备高锂离子电导率和对金属锂的本征稳定性,实现了前期预测。
【图文导读】
首先对体系进行理论分析,如图1所示,所有具备低形成能的新相均位于相图的边界处。作者通过结构预测发现了新的亚稳态Li2S−P2S5三元化合物,即Li5P3S10和Li5PS5,分别位于Li−P−S上方31.2和54.4 meV/原子处。
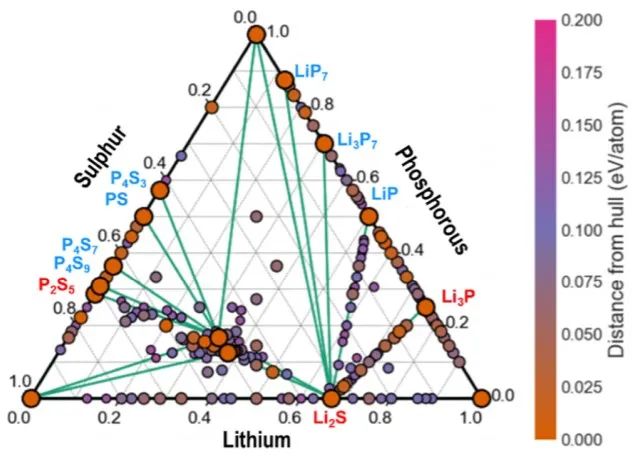
图1 Li−P−S三元相图及用DFT/ PBE随机结构分布. 顶点为Li/P/S纯相,边为二元相。
经过进一步分析,源自基础材料(Li2S和Li3P)的一些新结构在预测得到的三元基态结构中比较常见,即Li8P2S和Li5PS,分别是Li2S/Li3P的1:2和1:1的混合物(图2)。这表明两个相图中的顶点组分可能形成共生相。如图3所示,固溶体的衍射谱图都可以用一个基于Li2S反萤石晶体结构的模型进行对比测试,该晶体结构由硫化物阴离子的立方密堆积(ccp)组成。
由于S2−被较大的P3−取代,大量的Li3P使晶胞参数逐渐增加,当x=0.33, 0.50, 0.67 和0.75时,晶胞参数分别为5.83(8), 5.90(6), 5.96(3) 和5.98(6) Å。尽管在环境压力和温度下不同时的晶体结构不同,但晶格参数的变化依然遵循维加德定律。
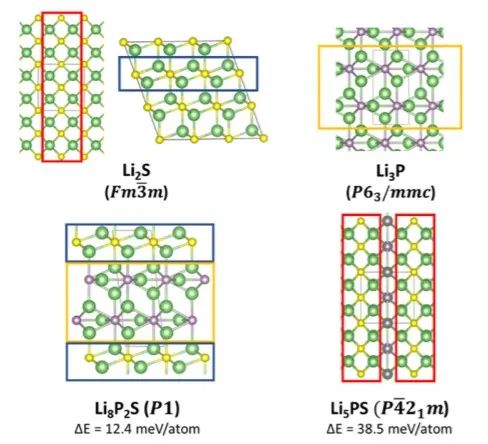
图2 基态结构Li2S和Li3P以及新预测的Li8P2S和Li5PS三元相。
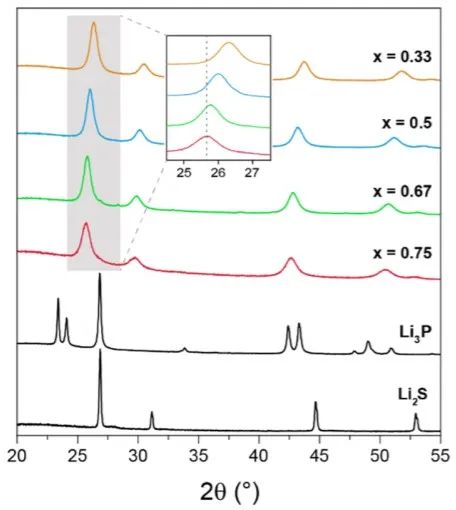
图3 xLi3P-(1-x)Li2S的XRD测试结果。 测得的XRD谱图与两种可能的晶体结构模型一致(图4a),在模型1的反萤石型衍生结构中,阴离子位置(Wyckoff 4a位点)具有P3−/S2−占位障碍,而额外的Li+则可被容纳在八面体空位中。模型2中,受限于电中性原则,额外的Li位点是不存在的。
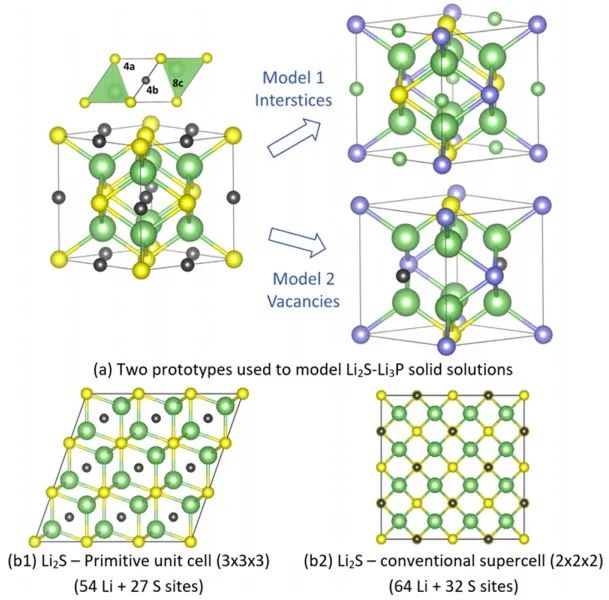
图4 (a) Li2S−Li3P固溶相的两种可能的结构模型;(b) 采用Li2S超胞作为缺陷构型起点的DFT计算结果。 图5a-b显示了x=0.5样品XRD和中子衍射共拟合结果。如图(a)所示,使用模型2对中子衍射数据精细拟合效果不佳,而基于模型1的拟合则成功地对中子衍射和同步辐射XRD谱图进行了建模。此外,XRD和NPD模式中观察到的反射较广,原则上这可以归因于高能球磨合成条件下产生的小晶粒尺寸或高应变。
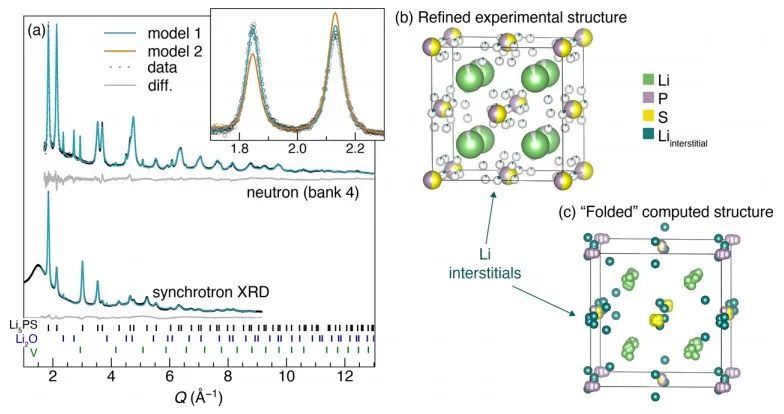
图5 对x=0.5样品(Li5PS)的同步辐射XRD和中子粉末衍射测试分析结果。(a)对XRD和中子衍射的Rietveld精修拟合结果,结构模型如图(b)所示,对应于模型1. (c) DFT弛豫计算,体系结构为x=0.5,其为折叠成单一的常规反萤石结构。 采用31P和6/7Li固态MAS NMR研究了固溶体的局域结构(图6)。7Li谱显示,对于纯的二元组分,2.2 ppm为Li2S信号,4.5和7.1 ppm则对应Li3P信号,确切的位移是由Li2S和Li3P的比值决定的。
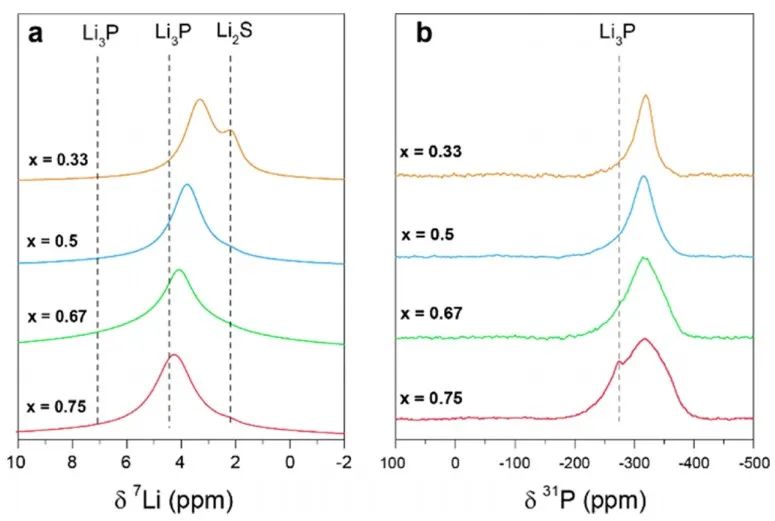
图6 室温下xLi3P-(1-x)Li2S的(a)7Li和(b)31P的MAS NMR测试结果。 在进行VT测量之前,作者首先评估了样品在高温下的稳定性。在光谱仪内加热样品到125°C, 7Li NMR谱线宽度减小(图7)。当样品在该温度下保持约1h时,线宽继续减小,这可能是样品和观测到的原子核分别在ns和μs时间尺度上的烧结所致。
相关时间与弛豫时间成正比,在不相关的三维运动中,阿伦尼乌斯行为十分显著,由此可计算得到体系活化能(图8)。对于固相体系,弛豫行为还表现出运动维数效应和相关效应,这会导致弛豫曲线的高、低温侧的表观活化能不同。
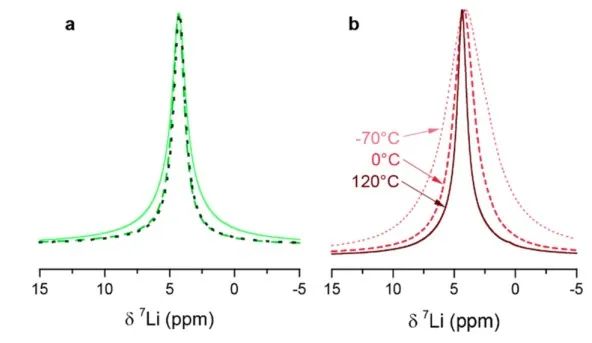
图7 (a) x=0.67样品直接球磨后的室温NMR谱(实线),在125℃退火后的1小时(虚线)、2小时(虚线). (b) x=0.75样品的温度相关性NMR谱。
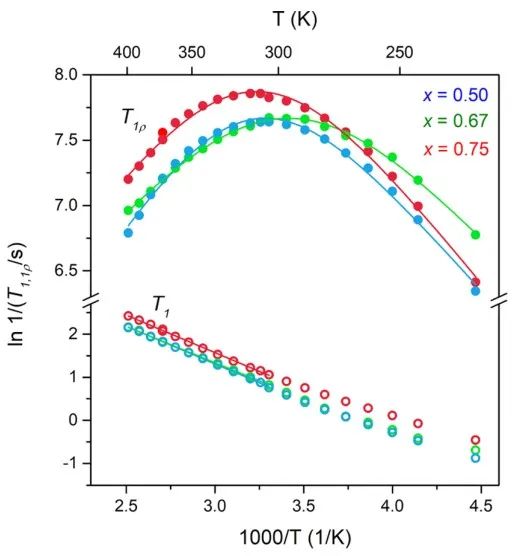
图8 xLi3P-(1−x)Li2S在-50-125℃下的7Li信号弛豫时间。 作者使用VT-EIS进一步探测锂离子迁移率,四个样品的室温阻抗如图9所示。所有样品的EIS图中都仅观察到一个半圆,因此无法区分晶粒(体积)和晶界的贡献。
对于x=0.5的样品,温度从100°C到RT和RT到−50°C的代表性VT-EIS图分别如图10a-b所示,所有样品的电导率都与温度呈线性关系。Arrhenius图给出的活化能Ea在0.2~0.15 eV之间,这与核磁共振弛豫测量得到的值一致,x=0.67样品的Ea最低。
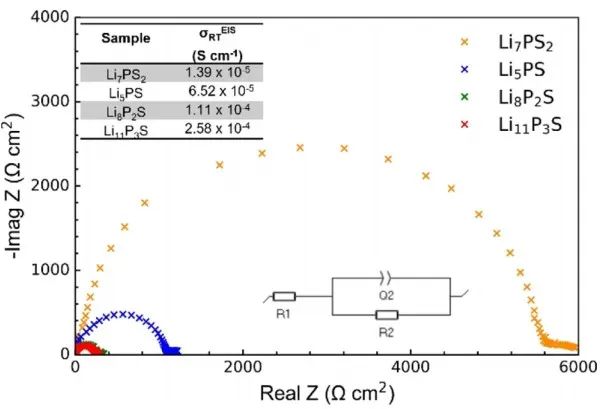
图9 xLi3P-(1-x)Li2S不同组分的Li|SE|Li对称电池的室温EIS图。
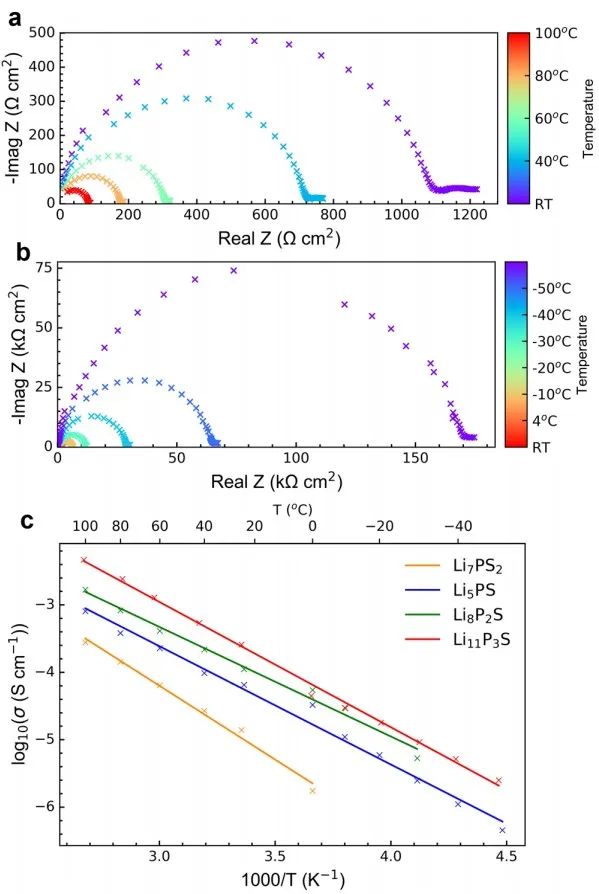
图10 x = 0.5样品在(a) 100°C至RT和(b) RT至−50°C时,Li|SE|Li对称电池的EIS图;(c) EIS-Arrhenius曲线显示了不同xLi3P-(1−x)Li2S组成的总离子电导率与温度的关系。 将通过DFT/GIPAW计算得到的x=0.67时7Li和31P NMR位移与实测结果进行了比较(图11),通过在298 K处的玻尔兹曼常数作为比例因子得到该测试结果。
基于以上结果,作者提出了一种基于反萤石的结构模型,其中S2-/P3-混合阴离子晶格为满占据状态,Li+(部分)占据四面体和八面体位点,空位则允许Li+跃迁。对所有成分进行类似的CE分析,得出Li2S−Li3P二元相图(图12)。
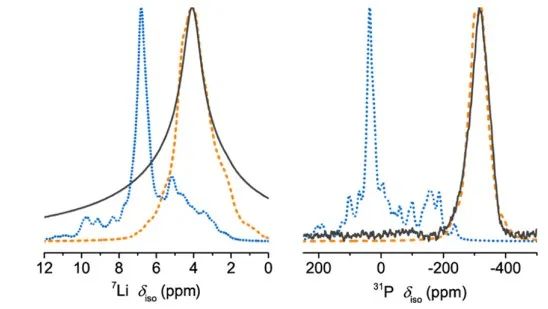
图11 Li8P2S三元相的预测7Li和31P NMR谱。
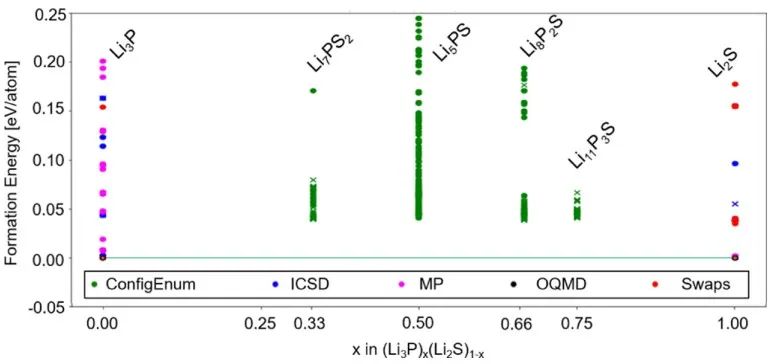
图12 Li3P−Li2S二元组分的预测结果(0 K)。
为了进一步验证预测的Li−P−S三元结构(图13),作者比较了计算的以及实测的7Li和31P NMR谱。预测的重心漂移分布随P含量的增加而变化愈发不明显。然而,在实测光谱中发现,这可能是因为计算没有考虑Li亚晶格的迁移所致。 如图14,作者还比较了离子电导率,并计算了相应的活化能(Ea),AIMD模拟结果与Li−P−S体系中已知的高电导率三元化合物进行了比较,当P含量越高时,由NMR和EIS得到的活化能越高。
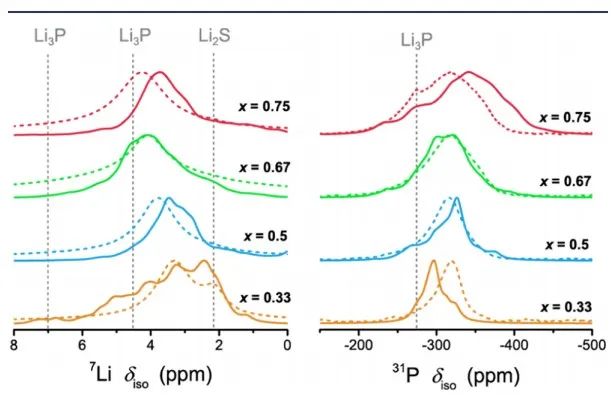
图13 Li-P-S的7Li和31P NMR谱的预测结果(实线)和实测结果(虚线)。
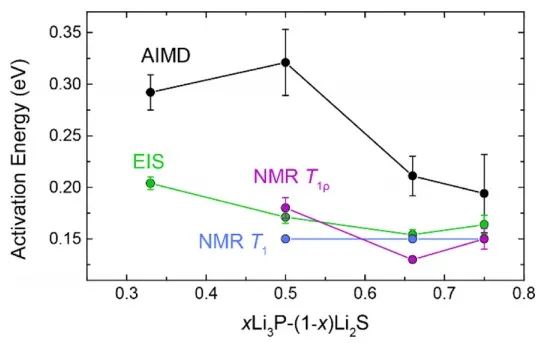
图14 预测和实验实测的 Li2S−Li3P活化能(Ea)。
【总结和展望】
本文报道了一种新的xLi3P−(1−x)Li2S固溶体的合成,以及结构解析和离子电导率测量,并以量子化学计算为支持。两种二元化合物高能球磨后,在0.39≤x≤0.75范围内形成反萤石结构类型的固溶体,最大的特点在于其整体为无序的阴离子晶格构型,四面体和八面体空洞被移动的Li+填充。这种材料具备高导电性和对金属锂本征的稳定性,因此可以被认为是潜在理想的固态电解质。该工作为开发高性能全固态电池提供了新的思路和见解。
审核编辑:刘清
-
labview用谐波合成方波2018-12-20 3112
-
三种频率合成方法2019-06-19 7695
-
求大佬分享一种有源滤波器合成方法2021-06-07 1235
-
电池材料之锗硅固溶体2009-11-09 861
-
一种视频合成方法2011-04-27 678
-
新型软化SPWM波形合成方法及谐波分析2011-09-22 1599
-
基于TPS54x60的电压轨生成方法2011-12-29 1855
-
一种多尺度多视点特性视图生成方法的研究和应用_谢冰2017-03-15 766
-
一种新颖的自动化攻击图生成方法_武健2017-03-19 863
-
基于证据价值的冲突证据合成方法2017-11-27 984
-
一种新的DEA公共权重生成方法2018-01-13 1161
-
一种在生成对抗性学习框架下的新颖的视频合成方法2020-04-09 2088
-
寻找形成固溶体合金成为材料设计的重要步骤2020-06-09 2110
-
基于鸿之微RESCU程序包的第一原理方法2023-06-02 1152
-
晶体知识——固溶体介绍2023-11-25 7811
全部0条评论

快来发表一下你的评论吧 !
