

利用可持续方法获得绿色能源的电催化分解水制氢
电子说
描述
【研究背景】
电催化分解水制氢是利用可持续方法获得绿色能源的有效策略。通常将Pt、Ir或Ru基金属分别用作析氢反应 (HER) 和析氧反应 (OER) 的电催化剂。然而,对于大规模电解水,上述催化剂达到200-500 mA cm-2的高电流密度仍然需要非常高的电压(1.8-2.4 V)。此外,贵金属元素的稀缺性、高成本和相对较低的电化学稳定性限制了其商业前景。因此,开发高成本效益、基于丰富的原材料、贵金属用量超低的催化剂非常关键。
具有不饱和位点的铁钴层状双氢氧化物 (FeCo-LDH) 催化剂能在碱性条件下同时实现HER和OER。为了满足低电压(<1.8 V)下大电流密度( 500 mA cm-2)的工业要求,必须进一步改进电解水催化剂。为了提高LDH的催化活性,一种经典的方法是通过打破催化剂表面结构的对称性,调整界面处的相互作用、配位环境和电子性质,促进电荷转移。金属-载体相互作用可以进一步提高本征催化活性。
500 mA cm-2)的工业要求,必须进一步改进电解水催化剂。为了提高LDH的催化活性,一种经典的方法是通过打破催化剂表面结构的对称性,调整界面处的相互作用、配位环境和电子性质,促进电荷转移。金属-载体相互作用可以进一步提高本征催化活性。
一种有效的策略是通过交替的Fe和Co原子排列,控制LDH生长;同时通过掺杂活性原子,构建多原子异质界面。活性金属原子进入LDH的骨架结构,破坏其有序原子排列的对称性,导致不饱和键断裂,并伴有局部电荷过剩,可能对催化活性产生积极影响。更重要的是,掺杂后得到的LDH以另一个原子作为新的活性位点,可以有效增加活性位点的数量,并诱导其杂原子界面处的电荷转移,从而调整反应吸附能,并调控分解水的性能。部分取代效应可以抑制工业反应条件下多杂原子界面的不稳定性。然而,如何打破多杂原子界面的对称结构,并构建基于LDH的稳定多原子异质界面,仍具有挑战性。
【成果简介】
武汉理工大学的木士春教授和南京晓庄学院的刘苏莉教授(共同通讯作者)在铁钴层状双氢氧化物 (Rux SACs@FeCo-LDH)上构建的Ru单原子催化剂在10和1000 mA cm-2的电流密度下表现出194和246 mV的极低析氧反应 (OER) 过电位,在1000 mA cm -2连续反应1000 h后,表现出极高的稳定性,其质量活度分别是Ru和FeCo-LDH的2倍和6倍。
【研究亮点】
1. 在1.52 V电压下,在电解水反应中可实现高达1000 mA cm-2的电流密度;
2. FeCo-LDH表面的结构对称性被破坏,在电催化条件下原位形成了类似于Ru-O-TM(Fe、Co 和 Ni)的纳米级化合物,促进O-O耦合。
【图文导读】
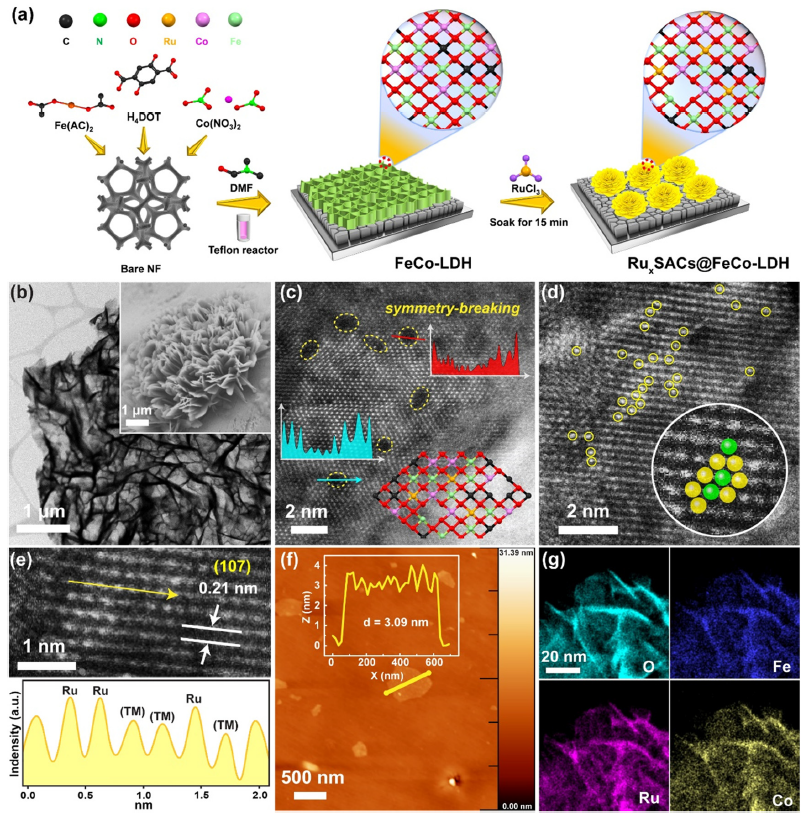
图1 (a) 水热合成电催化剂,(b) Ru1SACs@FeCo-LDH的TEM、FE-SEM和HAADF-STEM,(c)黄色虚线圈出的缺陷,(d) 明亮的黄色圆圈标记的点代表单原子Ru,(e)图像中白线之间的强度分布 (TM = Fe, Co),(f) AFM,(g) Ru1SACs@FeCo-LDH的元素映射图。
作者使用混合溶剂策略,用水热法合成RuxSACs@FeCo-LDH催化剂。如图1a所示,使用泡沫镍(NF)作为基底,先生长FeCo-LDH阵列,然后原位生长Ru单原子,从而打破了FeCo-LDH阵列原有的对称性,有利于催化反应。TEM和FE-SEM图像显示,Ru1SACs@FeCo-LDH仍然保持了由互连的超薄纳米片组成的均匀3D多孔结构(图 1b)。
如图1c所示,HAADF-STEM图像显示了催化剂表面对称性被破坏后的原子结构,这意味着在LDH表面形成了包含多种原子的界面,从而增加了活性表面积和活性位点数目。观察到的亮点(图 1d)对应于单个Ru原子,线性扫描分析进一步证明了Ru原子的孤立状态(图1e),但由于原子序数接近,无法区分Fe或Co原子。图1f中,Ru1SACs@FeCo-LDH纳米片厚度仅为 3.09 nm。能量色散 X 射线 (EDX) 光谱进一步证明了 Fe、Co 和 Ru 物种在FeCo-LDH纳米片上均匀分布(图 1g)。
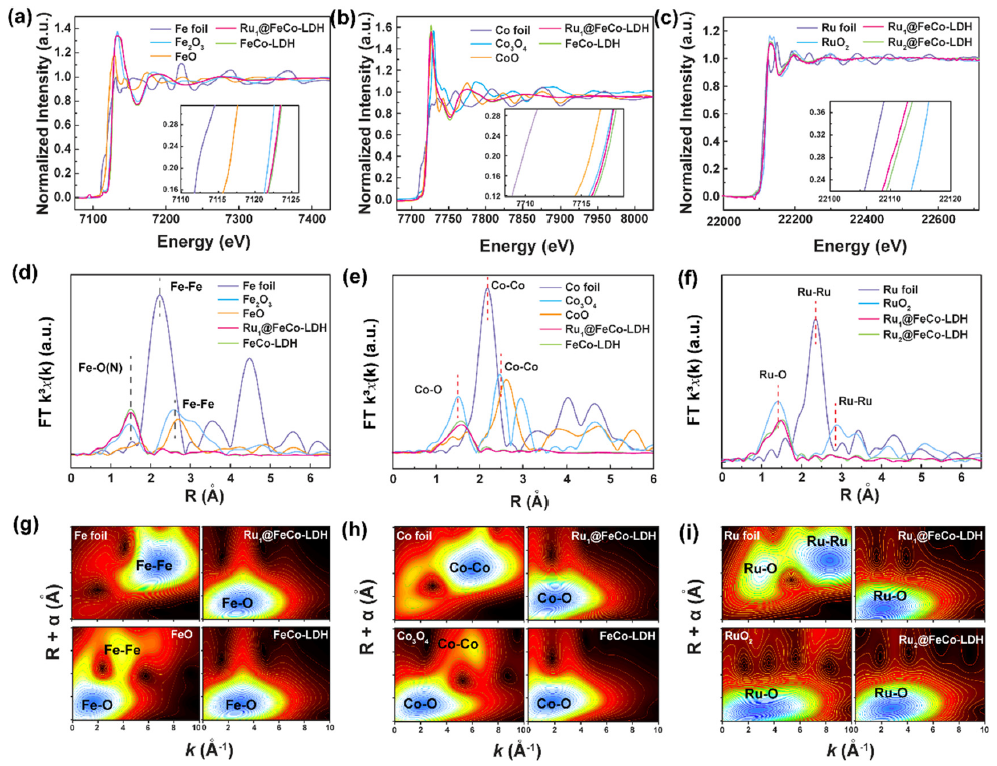
图2 Ru SACs@FeCo-LDH 的(a) Fe, (b) Co和 (c) Ru的K-边XANES光谱。(d) Fe 箔、Fe2O3、FeO、Ru1SACs@FeCo-LDH和FeCo-LDH的R空间傅里叶变换 (FT)。(e) Co箔、Co3O4、CoO、Ru1SACs@FeCo-LDH和FeCo-LDH的R空间FT。(f) Ru箔、RuO2和RuxSACs@FeCo-LDH的R空间FT。(g–i) Co、F和Ru 的 K 边 EXAFS 光谱的加权函数。
Ru1SACs@FeCo-LDH和FeCo-LDH样品的Fe XANES光谱在~7133.2 eV处显示出明显的前边缘峰(图2a),表明原子级分散的Fe物种呈+3价。与FeCo-LDH 相比,Ru1SACs@FeCo-LDH 显示出向较低光子能量的轻微转变,证实了Ru-O-Fe结构的形成。与FeCo-LDH相比,Ru1SACs@FeCo-LDH的Co K 边吸收边显示出相同的轻微负移,证明了价态介于+2和+3价之间的Co物种(图 2b)。图 2c表明,Ru的氧化态在0和+4价之间,并证明Ru原子带正电荷。如图2d所示,Fe元素的EXAFS光谱的傅里叶变换显示R = 1.5 Å 处的峰,对应于Fe-O。
对于Ru1SACs@FeCo-LDH,由于Fe和O在LDH结构中的结合,只能检测到Fe-O散射的信号,因此排除了Fe、Fe2O3的存在。Ru1SACs@FeCo-LDH的Co K边位于1.6 Å(图 2e)。图 2f中的FTR空间光谱表明,RuxSACs@FeCo-LDH 没有Ru-Ru键的散射峰,这反映了Ru1SACs@FeCo-LDH和Ru2SACs@FeCo-LDH中存在单原子分散的Ru。如图2g-i所示,Rux SACs@FeCo-LDH仅在 2.4 Å 处出现 Fe (Co, Ru)-O 键的特征峰,可以归属于原子 Fe ( Co, Ru)-O 物种。Fe-Fe、Co-Co和Ru-Ru配位的缺失消除了Ru簇存在的可能性。
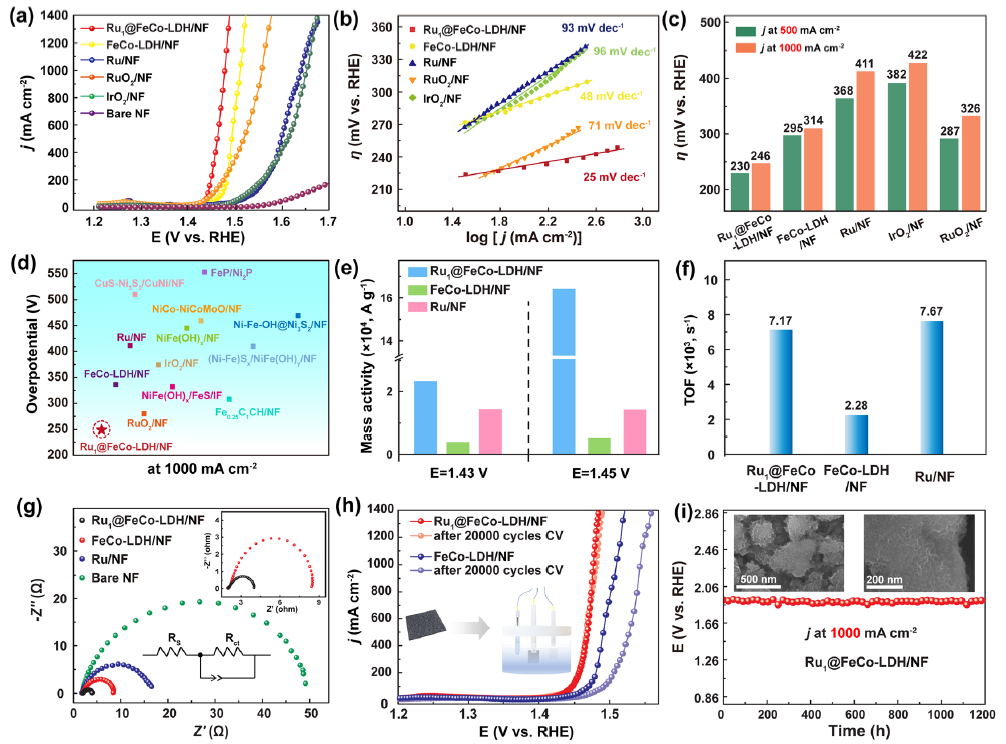
图3 (a) 1.0 M KOH中的极化曲线;(b)塔菲尔曲线;(c)在500 mA cm-2和 1 A cm-2下的过电位比较;(d) 在 1.0 M KOH 溶液中,与最近报道的1 A cm-2下的OER 催化剂过电位对比;(e) 不同过电位下的质量活性比较;(f) 不同电位下的TOF;(g) 奈奎斯特图;(h) Ru1SACs@FeCo-LDH和FeCo-LDH在20000圈CV循环前后的极化曲线;(i) Ru1SACs@FeCo-LDH在1 A cm-2电流密度下连续反应1200小时所需的电位随时间演化的计时电位曲线。
如图3a 所示,空白NF达到500 mA cm -2的电流密度需要439 mV的过电势。掺入单原子Ru后,Ru1SACs@FeCo-LDH表现出最高的OER活性。Ru1SACs@FeCo-LDH呈现出25 mV dec -1的最小Tafel 斜率(图3b)。图 3 c证明了 Ru1SACs@FeCo-LDH具有优异的OER催化活性。这比大多数先前报道的催化剂更好(图3d)。
对于Ru1SACs@FeCo-LDH,其在 200 mV 过电位下的质量活性(MA)分别是FeCo-LDH和Ru的6倍和2倍(图3e)。当过电位为200 mV时,转换频率 (TOF) 为7.17 × 10 3 s -1 (图 3f ) 。EIS证实了 Ru1SACs@FeCo-LDH 的快速反应动力学(图 3g)。如图3h和i所示, Ru1SACs@FeCo-LDH在1 A cm -2的电流密度下连续反应1200 h后,活性仅下降5%,表明其具有良好的稳定性。经历1200 h的OER后,HRTEM没有观察到明显的形貌变化。
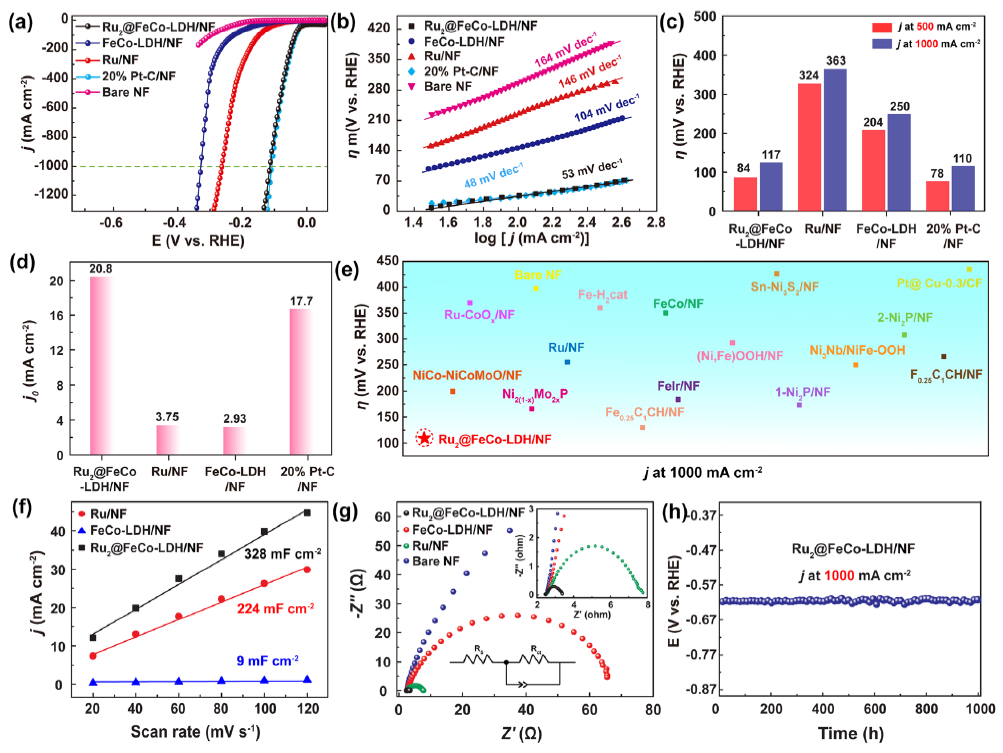
图4 (a) 1 M KOH中的极化曲线;(b)塔菲尔曲线;(c) 在 500 mA cm -2 和 1 A cm -2下电催化HER的过电位;(d) 交换电流密度 ( j 0 );(e) 在 1.0 M KOH 溶液中达到1 A cm -2的电流密度时,与最近报道的HER 催化剂的过电位对比;(f) C dl;(g) 奈奎斯特图;(h) Ru2SACs@FeCo-LDH在1 A cm -2下连续反应1000小时的计时电位曲线。
如图4a所示,Ru2SACs@FeCo-LDH 在碱性介质中表现出优异的HER活性, Tafel斜率低至53 mV dec -1(图 4b),接近商业 Pt/C 催化剂。如图4c所示,在500 mA cm -2和 1 A cm -2的电流密度下,过电势分别为 84 和 117 mV。与对照组催化剂相比,Ru2SACs@FeCo-LDH表现出最大的交换电流密度 ( j 0 ) (图 4d)。
Ru2SACs@FeCo-LDH的活性高于大多数报道的电催化剂(图 4e)。图 4f 表明,Ru2SACs@FeCo-LDH 显示出最大的Cdl和最小的R ct (图 4g),这表明快速的HER动力学来自增大的电化学表面积和显著提升的电荷转移能力。Ru2SACs@FeCo-LDH表现出优异的电化学稳定性(图 4h)。
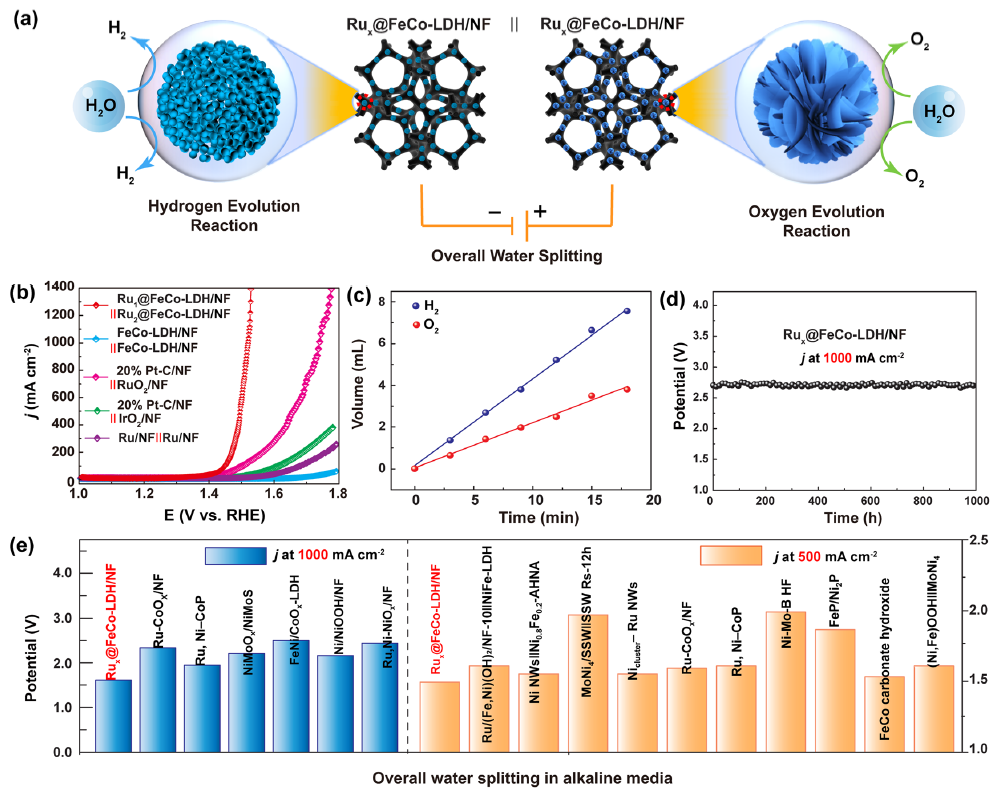
图5 (a) 全分解水装置;(b) 双功能RuxSACs@FeCo-LDH电催化剂的极化曲线;(c) Ru1SACs@FeCo-LDH的产气量;(d) RuxSACs@FeCo-LDH在1 A cm -2下连续反应1000 h的计时电位曲线;(e) RuxSACs@FeCo-LDH与报道的双功能电催化剂在500 mA cm-2和1 A cm-2下所需的电压对比。
作者组装了如图5a所示的碱性电解槽。与基于其他电催化剂的电解槽相比,在1.0 M KOH溶液中,该碱性电解槽表现出最佳的全分解水活性(图 5b)。从图5c可见,收集的H2和O2之比约为 2:1,法拉第效率接近100%,具有优异的耐久性(图 5d),可用于工业规模的全分解水催化剂(图 5e)。

图6 (a,b) RuSACs@FeCo-LDH和FeCo-LDH的Fe、Co和Ru活性位点的投影态密度。(c) FeCo-LDH和(d) Ru SACs@FeCo-LDH电催化OER过程中的自由能变化。(e)Δ GOOH*、 ΔGOH*、 ΔGO*和d带中心之间的比例关系。(f)作为d带中心函数的过电位火山图。(g) FeCo-LDH和Ru SACs@FeCo-LDH电催化OER的机制。
如图6a和b所示,Ru SACs@FeCo-LDH上,Co活性位点的d带中心比FeCo-LDH更接近费米能级,表明Ru SACs@FeCo-LDH在电化学反应过程中具有更强的电子供受能力。随着d带中心能级的提升,OER中间体的吸附能力增强,理论上可以提高FeCo-LDH的OER活性。基于FeCo-LDH的自由能图(图6c ), O2脱附是Co位点催化下的OER速控步骤。
将Ru引入FeCo-LDH后,Ru位点(图6d)增强了OER中间体的吸附能,即通过负移的d带中心优化了中间体的吸附自由能,从而促进了OER活性(图6e)。Ru掺杂降低了OER过电位(图 6f)。与 FeCo-LDH和Ru SACs@FeCo-LDH界面上的Co位点相比,活化能显著降低(图 6g)。
【总结与展望】
为了提升高电流密度下的全解水性能,该工作构建了强耦合单原子(Ru)改性的铁钴层状双氢氧化物(Ru SACs@FeCo-LDH),获得的RuSACs@FeCo-LDH在OER和全解水反应中表现出优异的电催化活性和稳定性。实验分析和DFT计算结果表明,由于FeCo-LDH被亲氧的Ru原子部分取代,打破了杂原子界面的长程有序,即打破了界面原子结构的对称性,进行了原子尺度重构,导致单原子Ru和FeCo-LDH之间形成了强耦合的肖特基界面。
审核编辑:刘清
-
CdS/ZnS 1D/2D异质结光催化剂用于高效光催化制氢2022-12-20 3286
-
EcoMat: 拓扑半金属用于电催化析氢反应的研究进展2022-12-13 2395
-
介绍三金属电催化剂对C2醇的电催化机理2022-10-20 4095
-
析氢反应(HER)电催化剂在电解装置的广泛应用2022-09-28 12163
-
一种高效评估复杂合金纳米团簇电催化析氢反应活性2021-06-18 3276
-
水氢机主要应用于工业电力、新能源汽车行业之中2021-05-19 951
-
电催化与电催化电极的原理及研究2021-02-10 3867
-
水氢机通过汽化催化重整及纯化等技术从甲醇水中提取氢2020-11-25 2170
-
“氢农场”助力太阳能光分解水制氢效率创下国际新高2020-03-26 2714
-
新型催化剂改善氢燃料使用 助力成为可持续替代品2020-03-25 3233
-
太阳能光催化分解水制氢2020-03-23 9241
-
光催化分解水制氢系统中的气体传感器应用2019-01-04 3558
全部0条评论

快来发表一下你的评论吧 !
