

DPO-TXO2如何读取数据用于结果分析
描述
鸿之微BDF(Beijing Density Functional)软件是一个独立完整、具有完全自主知识产权的量子化学计算软件包,也是国际上第一个基于现代密度泛函理论、能准确计算分子体系基态总能量的完全相对论密度泛函程序。
BDF的研发始于1993年,并于1997年正式命名。BDF对稀土、锕系、超重元素的计算结果一直被作为检验其他近似相对论方法的基准。BDF对重元素体系电子、分子结构的计算结果被后续20余个实验验证。
热激活延迟荧光(TADF)材料是继荧光材料和贵金属磷光材料之后发展起来的第三代纯有机延迟荧光材料,其典型的特征是较小的单三重态能隙(ΔES-T)和温度正相关性。
2012年,日本九州大学的Chiahaya Adachi课题组首次报道外量子效率(EQE)超过20%的4CzIPN 分子[ i ],该材料的单线态和三线态能级差几乎为0,在室温下(298 K)的这样的热扰动下激子完全能够从三线态再回到单线态而发射荧光,因此命名为TADF(Thermally activated delayed fluorescence)。
当S1与T1的激发都是HOMO-》LUMO特征,二者的能量差为2*K,K是HOMO与LUMO间的交换积分。随着HOMO与LUMO分离的增加,K会迅速减小。所以分离较大的时候,S1与T1 gap就较小,易于发生TADF需要的RISC。
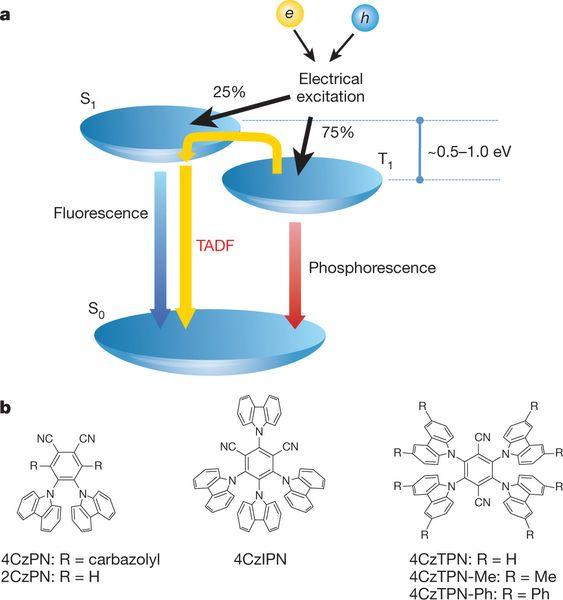
为了保证高效的RISC,TADF材料需要具有较小的单三重态能隙,对应其HOMO/LUMO的有效分离,因此,TADF材料一般采用给体(D)−受体(A)、D−A−D的结构以不同的给受体作用实现HOMO/LUMO分离,同时兼顾其跃迁振子强度。
不同给受体的电子特性、三重态能级、结构刚性及扭曲程度等均均会影响材料的△EST、振子强度、态密度、激子寿命等,最终反映在材料的光物理性能和对应OLED器件的光电性能上。
本专题将以一个典型的TADF分子DPO-TXO2为例,介绍如何计算结构优化、频率、单点能、激发能、自旋轨道耦合等。同时介绍如何读取数据用于结果分析,帮助用户深入了解BDF软件的使用。
一、结构优化和频率计算
1、生成结构优化和频率输入文件
在Device Studio中导入准备的分子结构DPO-TXO2.xyz得到如图1.1-1所示界面,选中 Simulator → BDF → BDF,在弹出的界面中设置参数。计算结构优化时计算类型选择Opt+Freq,方法、泛函、基组等选项用户可根据计算需要设置参数。例如Basic Settings面板设置为图1.1-2,SCF面板消除“Use MPEC+COSX”勾选(图1.1-3)、OPT 、Freq面板仍为默认值,之后点击 Generate files 即可生成对应计算的输入文件。生成的输入文件 bdf.inp参数部分如图1.1-4所示 ,此时Device Studio图形界面如图1.1-5所示。
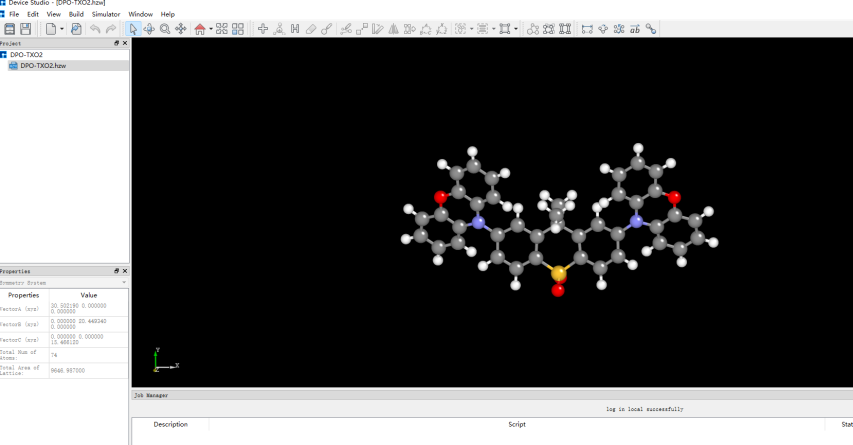
图1.1-1
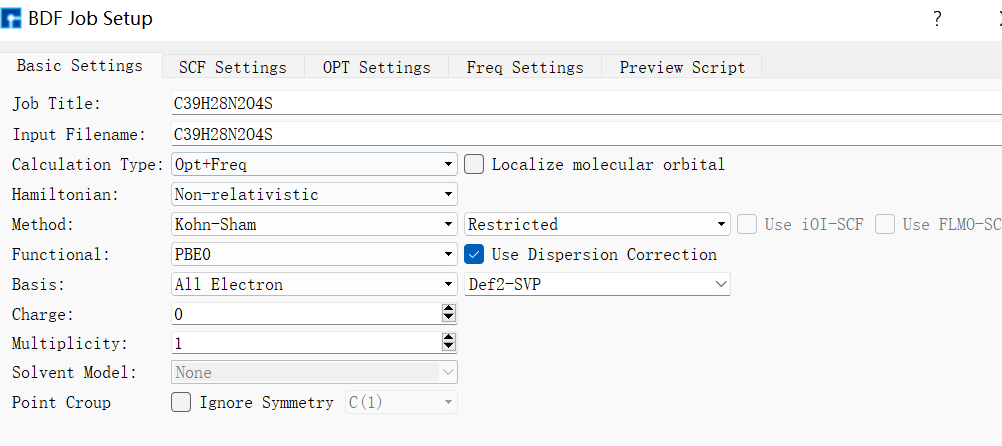
图1.1-2
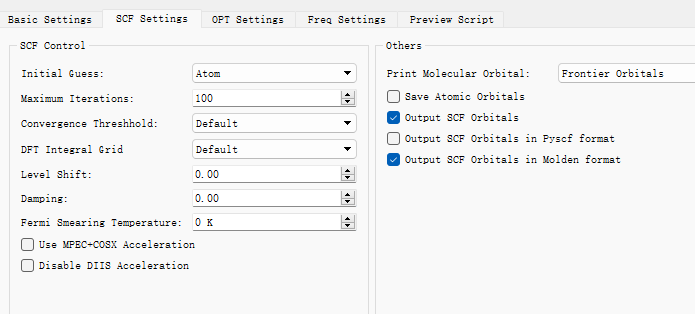
图1.1-3

图1.1-4
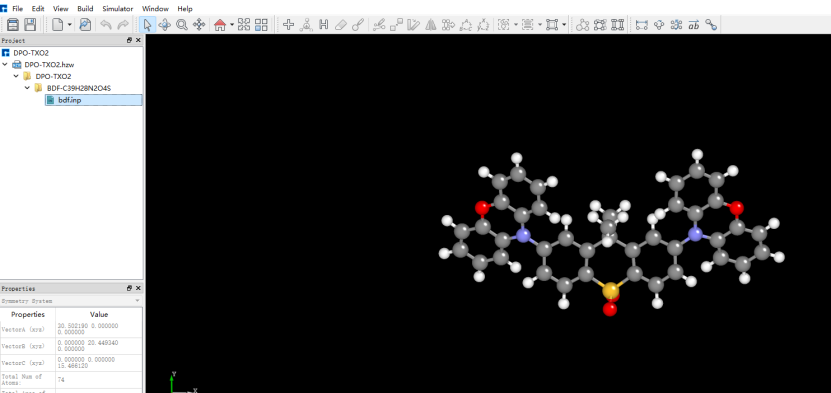
图1.1-5
备注:此处为保证结构优化和频率计算的条件相同,计算类型选择Opt+Freq,可以的单独做Opt计算或Freq计算。
2、BDF计算
在做BDF计算之前,需连接装有BDF的服务器,具体配置过程见鸿之微云操作指南。连接好服务器,在做计算之前,用户可根据需要打开输入文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中编辑或重新生成,再进行BDF计算。
在图1.1-5所示的界面中,选中 bdf.inp → 右击 → Run,在弹出的界面导入相应的脚本,点击Run提交作业,如图1.1-6。计算完成后点击下载按钮弹出计算结果界面如图1.1-7所示,选择.out结果文件,点击 Download下载。(提交作业操作为重复内容,在后面的计算中将不再赘述)
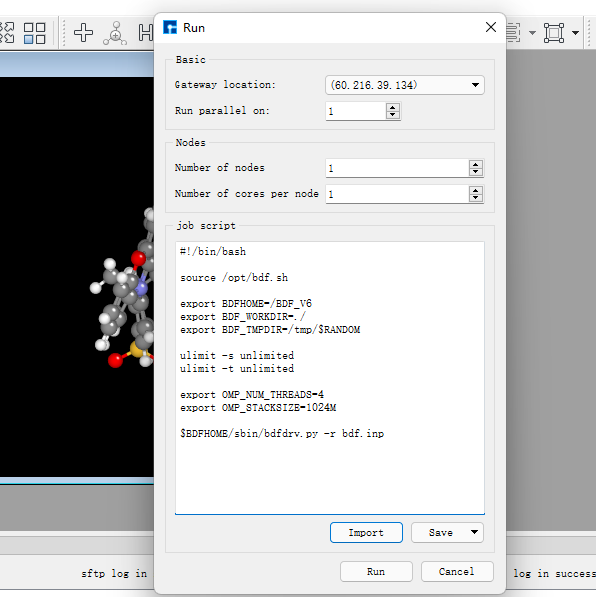
图1.1-6
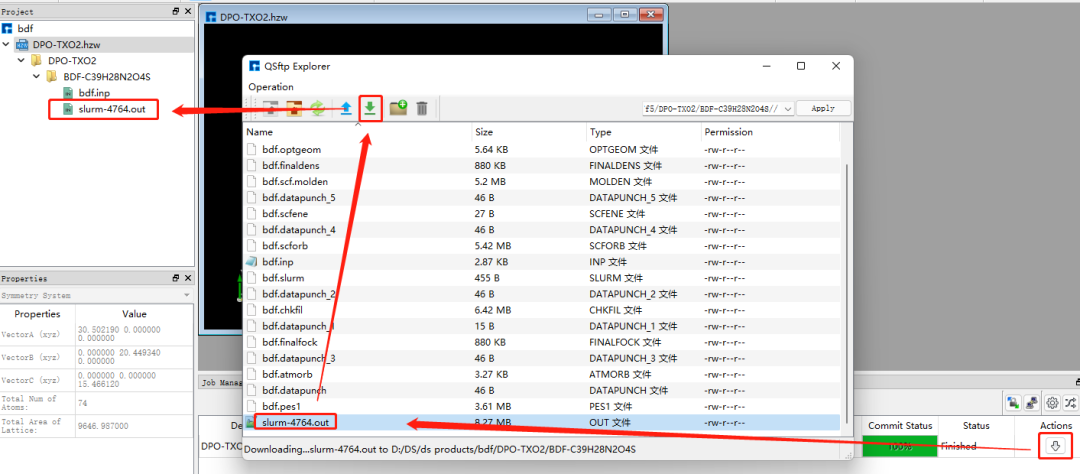
图1.1-7
3、结构优化结果分析
右击下载后的out文件,选择Open with/Open containing folder即可查看结果文件。找到如图1.1-8所示部分,当Geom.converge的4个值均为YES时,证明结构优化收敛。上方和下方分别为收敛的分子结构笛卡尔坐标和内坐标。优化后的坐标信息可以作为初始结构用于后续计算。

图1.1-8
检查频率,若不存在虚频证明结构稳定。
二、单点能计算

1、生成单点能输入文件
将优化后的坐标导入Device Studio,名字改为DPO-TXO2-sp.xyz,此时图形界面如图1.2-1。
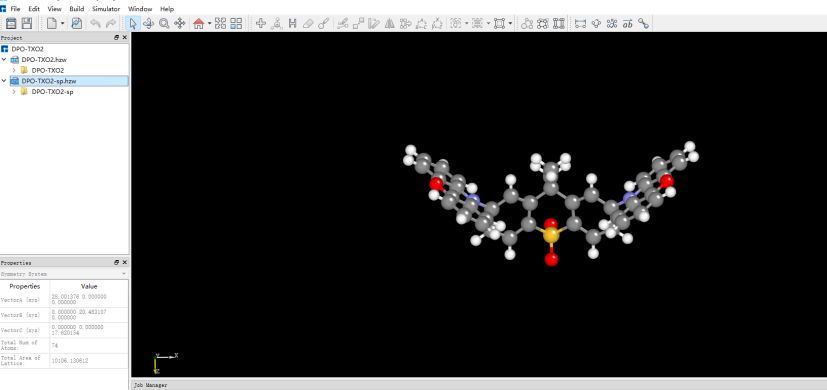
图1.2-1
选中 Simulator → BDF → BDF,在弹出的界面中计算类型选择Single Point(默认值),方法、泛函、基组等选项用户可根据计算需要设置参数。例如泛函选PBE0,基组Def2-TZVP,其他参数仍为默认值,之后点击 Generate files 即可生成对应计算的输入文件。生成的输入文件 bdf.inp参数部分如图1.2-2所示。
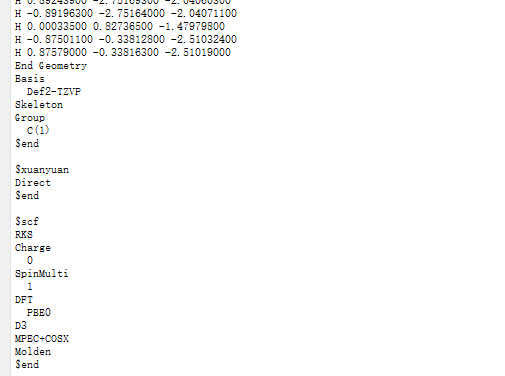
图1.2-2
2、BDF计算
同结构优化计算相同,连接好装有BDF的服务器后,选中 bdf.inp → 右击 → Run,检查脚本没有问题,点击Run提交作业。计算完成后点击下载按钮弹出计算结果,选择.out结果文件,点击 Download下载。
3、单点能结果分析
右击下载后的out文件,选择Open with/Open containing folder即可查看结果文件。找到E_tot为系统总能量(图1.2-3),E_tot=E_ele + E_nn,本例中系统总能量为-2310.04883102 Hartree。E_ele是电子能量,E_nn是原子核排斥能,E_1e是单电子能量,E_ne 是原子核对电子的吸引能,E_kin 是电子动能,E_ee 是双电子能,E_xc 是交换相关能。
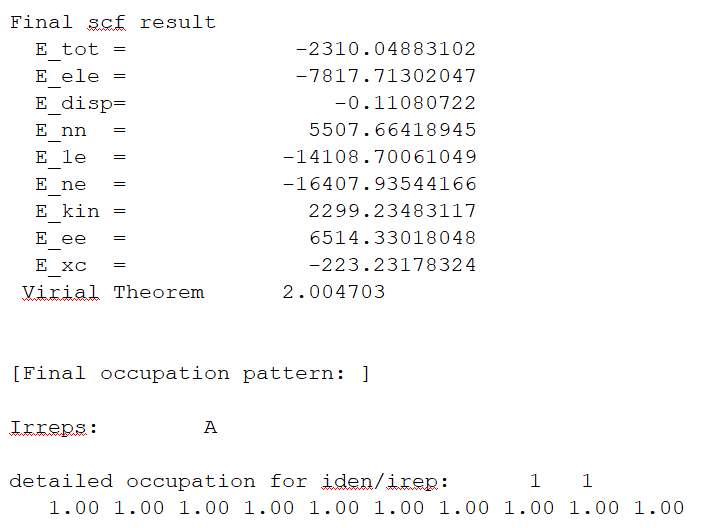
图1.2-3
下方为轨道的占据情况,以及轨道能、HOMO-LUMO gap等信息,如图1.2-4。HOMO为-5.358 eV,LUMO为-1.962 eV,HOMO-LUMO gap为3.396 eV,Irrep为不可约表示,代表分子轨道对称性,本例中HOMO、LUMO不可约表示序号均为A。

图1.2-4
最底部为Mulliken和Lowdin电荷布局、偶极矩信息。图1.2-5为部分截取。
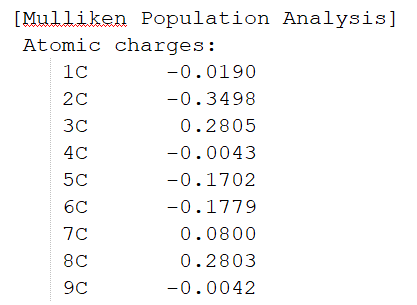
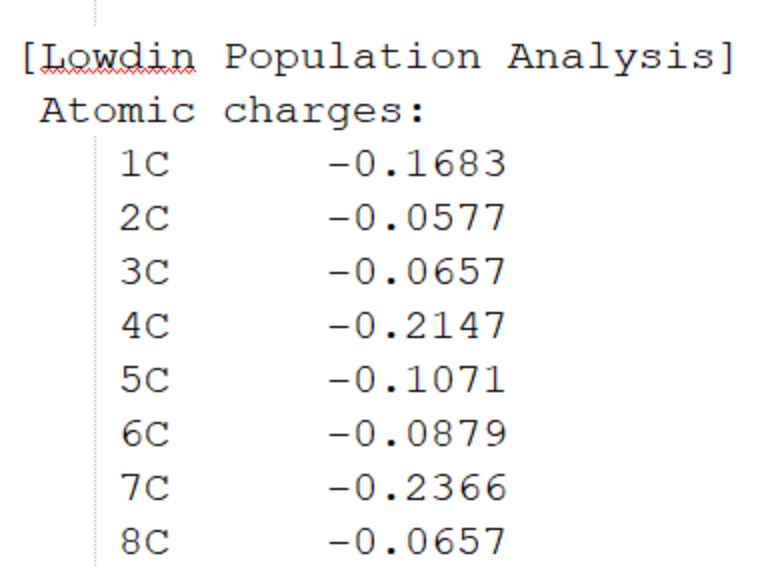

图1.2-5
4、查看HOMO轨道图
为了更清楚的了解电子结构,往往需要做前线分子轨道分析。目前发布的版本BDF2022A中还无法实现数据的后处理,HOMO、LUMO轨道图可以用第三方软件Multiwfn+VMD渲染,需要用到scf.molden文件,软件的使用方法在量化论坛有专门的帖子可以学习,此文不作涉及。
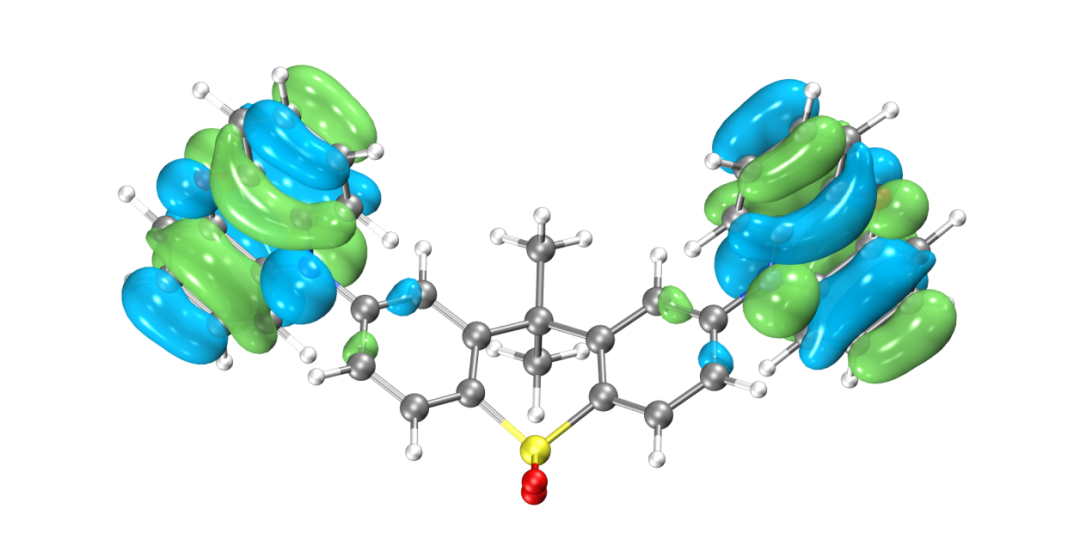
HOMO轨道分布图
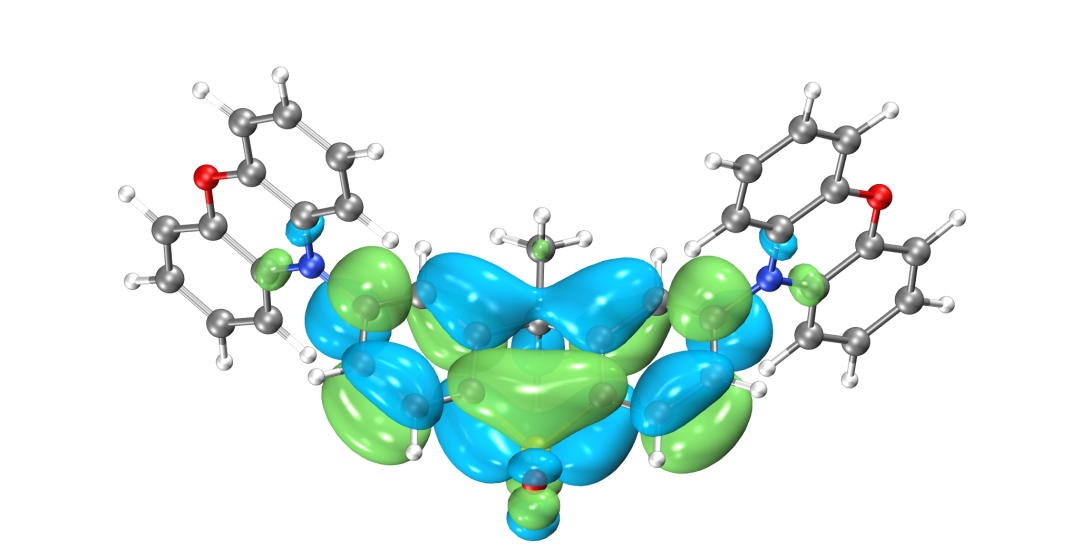
LUMO轨道分布图
得到的最高占据轨道(HOMO)与最低非占据轨道(LUMO)如图所示,由于两侧对称分布的吩恶嗪杂环是一个典型的给电子结构,而中心的磺酰化的四氢化萘是一个典型的吸电子的结构,因此整个分子是非常典型的D-A-D结构。可以看到HOMO轨道主要分布在两翼,LUMO轨道分布在中心,HOMO和LUMO轨道几乎没有重叠,符合TADF分子的电子结构特征。当然并不是所有HOMO和LUMO轨道分离的分子都具有TADF的光电特性,还需要满足S1和T1激发都是HOMO-》LUMO轨道跃迁才行,因此我们可以进一步用BDF软件计算该分子的激发态电子结构。
-
长期收购Tektronix泰克DPO5204/DPO5404B 2G数字示波器2021-12-21 427
-
美国原装Tektronix泰克DPO3012, DPO3014, DPO3052 数字示波器2021-09-08 638
-
出售DPO4104B 出售DPO4104B2021-08-15 642
-
DPO3054 求购二手 DPO30142021-01-31 955
-
佰福达回收示波器DPO5104B泰克DPO5104B2020-11-11 960
-
DPO7254回收DPO7254C泰克示波器2020-08-20 845
-
收购示波器DPO4034B二手DPO4034B2020-04-20 510
-
高价回收泰克/Tektronix DPO3034 DPO3032报价、价格2020-03-02 867
-
DPO4104B 示波器美国泰克DPO MSO4000B系列数字示波器2018-12-11 2024
-
DPO4054 DPO4054数字荧光示波器DPO4054 DPO4054数字荧光示波器DPO4054 DPO4054数字荧光示波器2018-11-01 1302
-
Agilent DPO4104B DPO4104B 示波器DPO4104B2018-10-24 1224
-
DPO75902SX回收|二手DPO75902SX泰克示波器2016-04-21 2375
-
DPO7000系列和DSA/DPO70000/DPO70002010-11-03 1810
-
网络分析仪数据读取工具软件2008-04-21 1173
全部0条评论

快来发表一下你的评论吧 !
