

Co掺杂实现普鲁士蓝自修复助力长寿命钾离子电池
电池
描述
01 导读
电极材料形貌的保持有助于延长电池的使用寿命。然而熵增是一个自发过程,会导致电池中电极材料形貌的剧烈变化并引发一系列副反应,如体积膨胀、界面衰退和枝晶生长。以上现象不可避免地破坏了电极结构形态并减少了离子存储活性位点,导致循环稳定性变差。已有研究人员提出自修复策略,以保持电极结构和电化学性能。然而,大部分自修复策略主要是作为保护层,而非活性储能材料,且只能应用在负极上。
02 成果背景
鉴于此,香港城市大学张文军教授与澳门大学洪果教授(共同通讯作者)等人选择性地在六氰化铁(FeHCF)结构中掺入微量的钴(Co),将其用作钾离子电池正极,基于"电化学驱动的溶解-结晶"机制的自愈效应,可以实现优异的循环性能。
(1)元素掺杂诱导材料自修复,使得钾离子电池中PBA正极的形貌得以恢复;
(2)实验表征和理论计算表明,微量的钴掺杂可以减速结晶过程并恢复裂纹区域,以确保PBA正极的立方结构,Co的掺入可以有效地调整FeLS-C八面体的电反应性,利于材料实现自修复。
04 核心内容解读
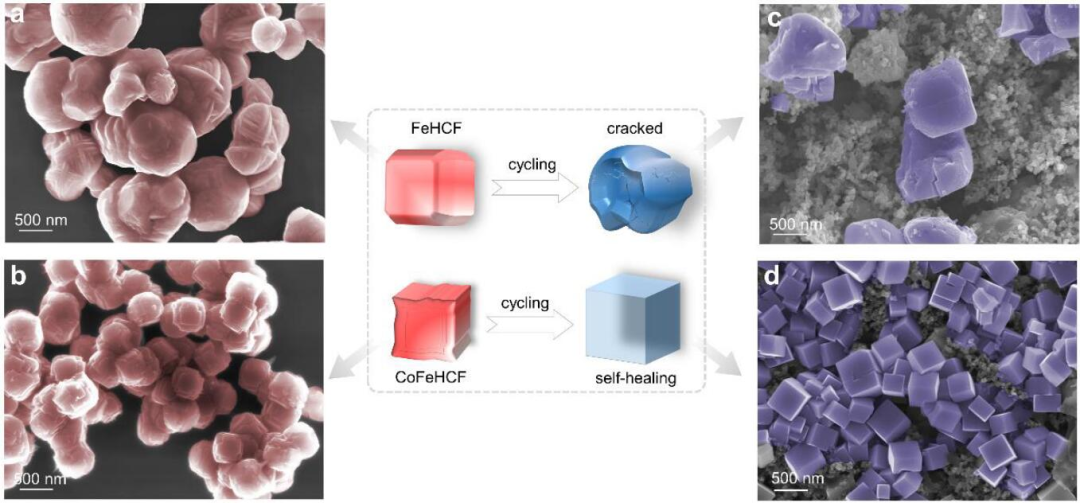
图1 (a)FeHCF和(b)CoFeHCF的SEM图像,(c)FeHCF和(d)CoFeHCF经过50次循环后的SEM图像。@ wiley
首先通过水热法合成了FeHCF和CoFeHCF,如SEM图像所示(图1a-b),FeHCF为不规则的纳米颗粒,而CoFeHCF则为准立方体结构。充放电50次后,FeHCF结构发生坍塌,存在明显的裂纹,而CoFeHCF呈现完美的立方体结构(图1c-d)。与最初的样品相比,FeHCF表现出明显的形态退化,而CoFeHCF显示出自修复效应使得形貌得以保持。
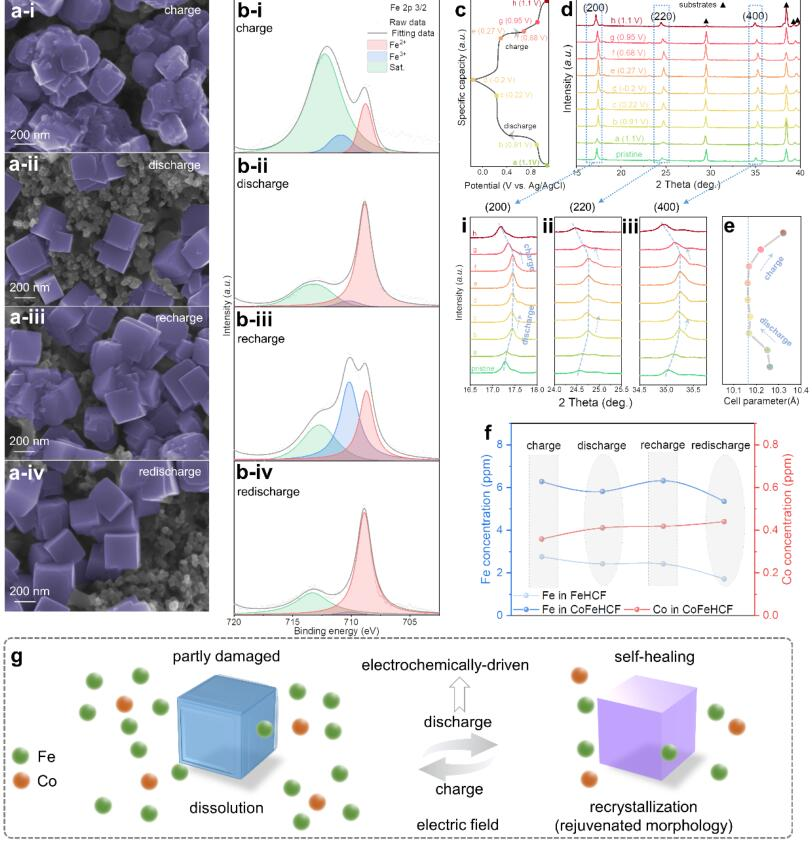
图2 (a) 不同充放电状态下CoFeHCF电极的SEM图像和(b)XPS光谱;(c)标记的充电-放电曲线;(d)XRD图案变化和(e)相应的电池参数;(f) CoFeHCF电极在第21和第22个周期时的电解质中铁/钴浓度变化;(g) "电化学驱动的溶解-结晶过程"的示意图。@ wiley
随后探究了充放电过程中CoFeHCF的形态变化(图2a)。CoFeHCF电极在第21次充电过程后发现边界较为丰富(2a-i),在第21次放电过程后转变为完美的立方体结构(2a-ii),表明放电状态下可逆的自修复过程。此外,在下一个循环周期时(2a-iii和iv),形态保持相同的趋势,显示出形态的周期性变化。
CoFeHCF的XPS结果显示(图2b),Fe2+的特征峰在充电状态(2b-i和iii)下变得较弱,在放电状态(2b-ii和iv)下变得较强。此外,发现Fe3+的比例在充电(2b-i和iii)和放电(2b-ii和iv)过程后分别增加和减少,表明存在一个可逆的氧化还原反应。CoFeHCF立体框架是由二价铁的高还原性元素驱动的,而不是由其三价对应物驱动的,这表明还原性电场促成了自愈现象。
作者通过连续充电/放电状态下的XRD图谱(图2c)来探究电场下晶体结构的演变过程。在XRD图中没有观察到新的特征峰(图2d),因此,固溶过程没有引起相变。主要衍射峰((200),(220)和(400))在最初的放电过程(i-iii)中发生明显移动,表明由于K+的插层使得晶格参数减小。当进一步放电到0.22V以下时,晶格参数保持不变,并在在充电过程中移回较小的数值。如图2e所示,在整个周期中,晶格膨胀和收缩率始终低于1.0%。这种可逆过程有利于保持结构和电化学稳定性,以实现自修复。
通过ICP-MS对充电-放电过程中电解液中铁和钴的浓度进行表征(图2f)。溶解在CoFeHCF中的Co浓度很小而且几乎恒定,而Fe则以周期性振动的趋势溶解。FeHCF在电解液中的铁浓度不断下降,这表明结晶过程的增强是随机和不可控的(图1c)。CoFeHCF的电解液中铁浓度的周期性波动可能是由电解液中稳定的钴浓度控制的,这与CoFeHCF的SEM和XPS的周期性变化相一致(图2a-b)。因此,在充电过程中,铁溶解到电解质中,并在放电过程中,在钴的捕获下,可以可逆地适当再结晶,有助于立方体形态的自修复。热力学在快速的充放电过程中不可能发挥主导作用,这种现象只能通过电场下的动力学来确定,即PBA电极的周期性形态变化的"电化学驱动溶解-结晶过程"(图2g)。
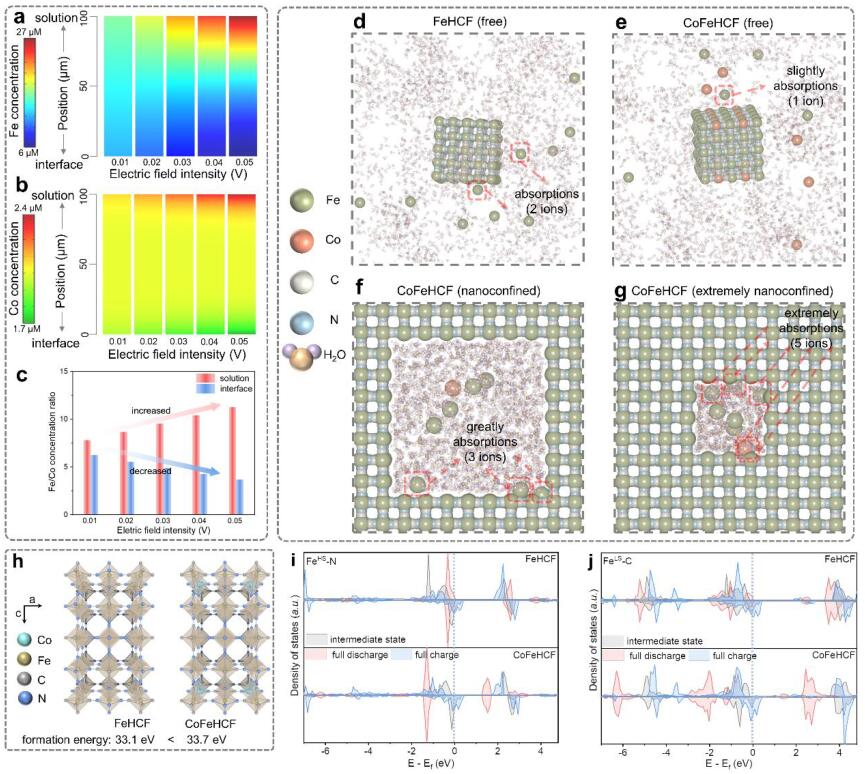
图3 采用COMSOL Multiphysics对增加电场下CoFeHCF电极的(a)Fe和(b)Co浓度在电解质中空间变化进行建模;(c) CoFeHCF电极的溶液和界面中的铁/钴浓度比的变化;(d)自由状态下的FeHCF和(e)CoFeHCF以及(f)纳米封闭和(g)极端纳米封闭条件下的CoFeHCF的三维快照的MD模拟;(h)对FeHCF(左)和FeCoHCF(右)的晶体结构和相应的形成能进行DFT计算;(i)FeLS-C和(j)FeHS-N在不同充电-放电状态下的Fe 3d的PDOS。@ wiley
离子浓度分布是溶解-结晶过程中的关键参数,因此通过COMSOL多物理场建模,模拟了电场增强时的离子空间变化(图3a-b)。电场增强时,铁离子和钴离子都表现出从界面到溶液的浓度梯度逐渐加强的特点。此外,铁/钴浓度比在溶液中增加,在界面上减少(图3c),这意味着钴在再结晶过程中起着主导作用。
采用分子动力学(MD)模拟分析了电极和电解质之间的界面相互作用。如图3d-e所示,FeHCF中的两个Fe离子在电极表面周围被吸收,而CoFeHCF中只有一个Fe离子被吸收,这是由于电解质中Co对Fe的吸引。电解液中溶解的Fe在FeHCF中显示出较快的反应速率,导致严重的颗粒聚集,而电解液中溶解的Co可以捕获Fe并减缓反应速率,从而促进CoFeHCF的结晶。
此外,在初始状态下进行了纳米封闭的MD模拟,以模拟缺陷区域的自修复选择性。在纳米封闭模式下,共有三个铁离子在电极周围被显著吸收(图3f),但在自由状态下只有一个铁离子被吸收(图3e)。表明结晶发生在裂纹区,而非未受损区。Co协助自修复可以快速处理初始阶段的缺陷区,从而抑制晶体的快速生长,促进再结晶过程。因此,反应速度较慢的Fe倾向于附着在由K离子嵌入/脱出引起的裂纹区域),而不是随机区域,引起自修复现象。
DFT计算表明,CoFeHCF具有比FeHCF更大的形成能,在掺入Co后呈现出较慢的结晶速度(图3h)。图3i中FeLS-C的部分态密度(PDOS)具有几乎完美对称的电子密度分布,在充电过程中逐渐接近费米能级,而图3j中FeHS-N的对应部分则远离了费米能级。随着Co的引入,FeLS-C八面体表现出比FeHS-N更高的反应性,在完全放电状态下提供了一个完整的立方体框架,具有良好的电传输特性。此外,如图3j所示,与FeHCF相比,Co的引入明显改变了CoFeHCF中FeLS-C的PDOS分布。
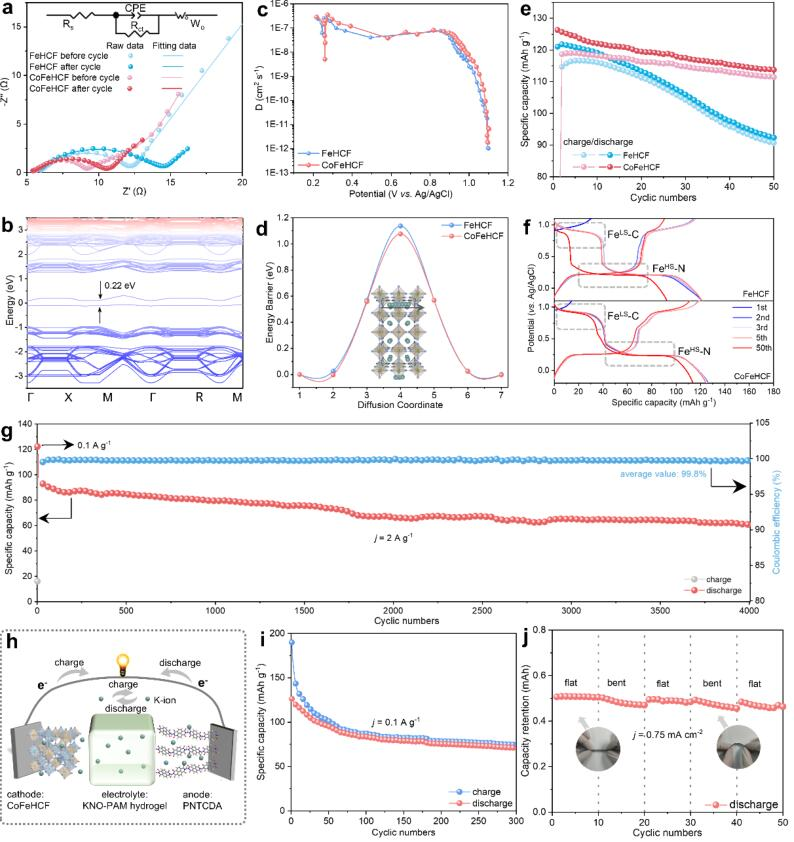
图4 (a) FeHCF和CoFeHCF在10个循环前后的EIS谱;(b) CoFeHCF的自旋极化电子带结构;(c) 从GITT曲线计算的扩散系数;(d) K离子扩散能垒和相应的CoFeHCF迁移路径的计算模型;(e) 在电流密度为0.1 A g-1时,FeHCF和CoFeHCF的循环性能;(f,e)第1至50个循环的对应GCD曲线;(g) CoFeHCF在电流密度为2 A g-1时的长期循环性能;(h) 全水基KIBs的示意图和(i)电流密度为0.1 A g-1时的循环性能;(j) 柔性器件在不同弯折状态下的循环性能。@ wiley
图4a中的电化学阻抗光谱(EIS)显示,与FeHCF相比,CoFeHCF电极的电荷转移阻抗(Rct)明显较低。在图4b中,可以观察到CoFeHCF的带隙(0.22 eV)比FeHCF的带隙(2.31 eV)小得多,表明其准金属态的特性。图4c显示CoFeHCF电极的扩散系数高于FeHCF。
图4d显示CoFeHCF比FeHCF(1.14 eV)具有更小的K离子迁移能垒(1.08 eV)。如图4e所示,CoFeHCF电极表现出119 mAh g-1的初始可逆比容量,而FeHCF为114 mAh g-1。50个循环后,CoFeHCF电极保持了111 mAh g-1的高容量,而FeHCF的容量下降到91 mAh g-1。如图4g所示,CoFeHCF电极在低浓度电解质中4000次循环后,在2 A g-1时表现出61 mAh g-1的容量,平均库仑效率为99.8%。
使用KNO3-聚丙烯酰胺(KNO-PAM)水凝胶电解质和1,4,5,8-萘四甲酸二酐(PNTCDA)负极构建了水系全电池(图4h)。全电池在300次循环后,在0.1A g-1时显示出71 mAh g-1的放电容量(图4i)。且全电池可以在反复弯曲的状态下为电子装置供电,且容量得到了良好的保持(图4j)。
05 成果启示
本工作通过实验以及Multiphysics建模、MD模拟和DFT计算表明,在PBA晶格中掺杂Co可以缓解晶体表面的Fe迁移,从而恢复晶体形态。相应的"电化学驱动的溶解-再结晶过程"表明,可逆的再结晶是由电场控制的,而Co的掺入可以有效地调整FeLS-C八面体的电反应性,从而促进自愈现象。这种掺杂Co的策略可以提高普鲁士蓝类似物的使用寿命,自修复普鲁士蓝类似物有望成为新一代柔性水系钾离子电池电极材料。
审核编辑:刘清
-
宽温长寿命贴片电阻的工业级探索2026-01-20 662
-
蔚来与宁德时代在北京签署框架协议,推动长寿命电池研发创新2024-03-15 1434
-
蔚来提出长寿命电池解决方案,目标实现电池使用15年2024-03-14 1809
-
长寿命桥梁设计2020-10-14 1868
-
哪家电池更耐用?超威长寿命电池揭开背后真相!2020-07-07 7022
-
长寿命锌基液流电池用复合离子传导膜研究取得新进展2020-03-18 2759
-
香港中文卢怡君Nature Mater:高效率长寿命的有机-氧电池2019-04-01 4687
-
Digi采用电池供电长寿命无线传感器XBee2018-10-29 1466
-
低成本高性能钾离子全电池德国诞生2017-01-09 3594
-
华为电池获突破:首个业界高温长寿命石墨烯基锂离子电池2016-12-01 1174
-
长寿命NTC热敏电阻2013-07-27 3808
-
日立研发长效电池 最长寿命10年2010-04-06 873
-
怎么对烙铁头进行的正确维护及延长寿命2010-02-27 2752
-
惠普Sonata锂离子长效电池超长寿命电池开始销售2009-11-10 742
全部0条评论

快来发表一下你的评论吧 !
