

一种相变电解质(PCE)
描述
一、 全文概要
Li+溶剂化结构(LSS)被认为是决定锂金属电池电化学性能的决定性因素。来自北京航天航空大学的李彬团队提出了一种相变电解质(PCE),其LSS可以通过改变电解质的物理状态来进行调节。PCE的主要溶剂是十二烷二酸二甲酯(DDCA),在一系列溶剂中脱颖而出,具有优异的综合性能。
PCE在固态和液态下表现出高离子电导率、锂迁移数和宽电化学稳定性窗口。此外,所得PCE的LSS可以根据其物理状态在溶剂分离离子对(SSIP)和接触离子对(CIP)结构之间进行可逆转换。这一特性使PCE在抑制活性负极材料的锂枝晶生长和溶解方面优于传统的液态电解质,尤其是在不同的温度下。组装的Li-LiMn2O4全电池表现出出色的电化学性能、高库仑效率和容量保持能力。
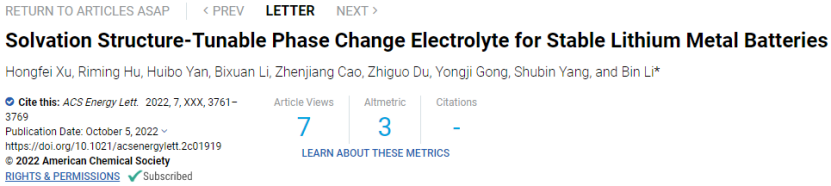
二、 研究亮点
1、在这项工作中,筛选了一系列具有较高熔点(>10°C)的电解质溶剂,其中十二烷二酸二甲酯(DDCA)溶剂基电解质(PCE)表现出优异的整体性能,包括高氧化稳定性,与锂金属和离子电导率。
2、利用拉曼光谱和分子动力学(MD)模拟,发现LSS还可以根据PCE的物理状态在SSIP和CIP之间转换。LSS的温度依赖性转变有助于调节Li+传导机制,即使在其固态下也可以获得相对较高的离子电导率。
3、得益于PCE的高迁移数和负极侧的有机-无机杂化SEI,在液态PCE中观察到无枝晶沉积层。
4、具有CIP结构的固态PCE减少了游离溶剂分子,并提供了天然的物理屏障来抑制过渡金属离子的穿梭。结合PCE的相变特性及其可转换的LSS,提出了一种精心设计的循环协议,并在LiLiMn2O4(LMO)系统中进行了测试。缓解了LMO正极中长期存在的锰离子溶解问题,获得了稳定的LMO电池性能。
三、正文导读
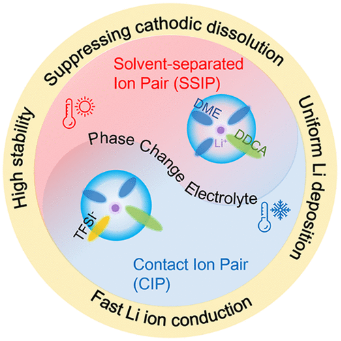
获得的PCE(1.3 M LiTFSI在DDCA:DME=3:1 vol%)在22°C的熔点下具有相变特征。高于或低于该温度的温度会导致物理状态(液态固态)的可逆变化(图1)
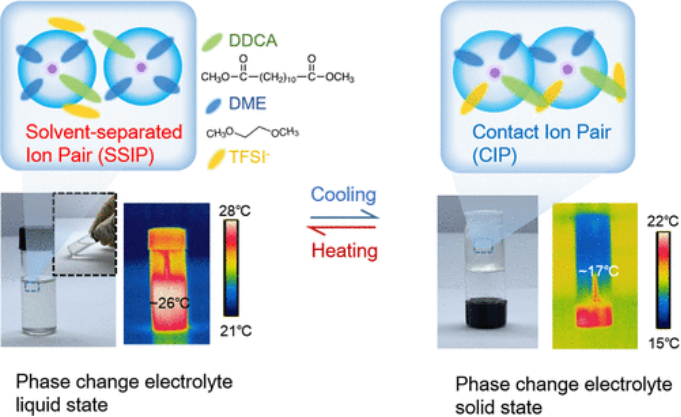
【图1】制备的PCE和相应溶剂化结构的示意图。照片和红外图像显示了固态和液态PCE的物理状态和整体温度。上面的方框显示了两种状态下相应的溶剂化结构。
在设计常规电解质时,通常会排除具有高熔点的溶剂,因为当溶剂固化时离子电导率会降低。然而,没有人研究过一种电解质的LSS在其不同物理状态下的变化,LSS与物理状态耦合的应用仍有待充分探索。选择并筛选了五种有机溶剂(熔点>15°C),即十二烷二酸二甲酯(DDCA)、丁二腈(SN)、碳酸亚乙酯(EC)、环丁砜(SF)和十八烷(烷烃C18)。
随着温度降至熔点以下,它们都从液态转变为固态。 它们都在略低于或高于30°C时具有适度的相变点。除烷烃C18外,其他四种溶剂对锂盐均表现出优异的溶解性(以LiTFSI为代表)。通过分子轨道计算证明了其余溶剂的抗氧化性。热力学上,电解质组分的最高占据分子轨道(HOMO)代表了它们的电子亲和性,在一定程度上可以反映高压相容性。
如图2a所示,DDCA、EC和SN表现出相对较低的HOMO,这应该具有更高的抗氧化性。对溶剂LiTFSI系统进行了线性扫描伏安法(LSV)。在四个样品中,DDCALiTFSI表现出最好的抗氧化性(~5 V)。与分子能级结果一致,EC还具有4.65 V的良好抗氧化性。SN已被视为液态电解质中的高压添加剂。
然而,由于SN与锂金属之间存在严重的氧化还原副反应,具有最宽能级窗口的SN-LiTFSI显示出低抗氧化能力和不稳定的LSV曲线。离子电导率是在20和35℃下测量的。由于SF的熔点相对较低和SN的塑性晶体性质,它们在20°C时表现出比EC-LiTFSI(3.3×10-7 S cm-1)高得多的离子电导率。
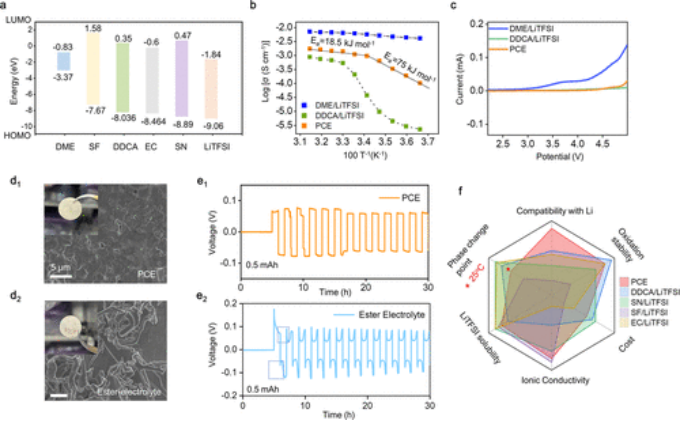
【图2】PCE的表征和评估。(a)PCE和DDCA-LiTFSI、DME-LiTFSI的离子电导率在0到45°C的温度依赖性。(b)相变点接近室温的所选溶剂的HOMO/LUMO能级。(c)这些选定样品在1 mV s-1的扫描速率下的LSV曲线。(d)PCE-L(d1)和酯基电解质(d2)中锂生长形态的SEM图像。(e)对称电池在选定电解质中的恒电流循环,0.5 mA cm-2,1h。(f)雷达图显示了所选相变电解质的评估标准。
DDCA具有优异的抗氧化性。为了进一步提高DDCA-LiTFSI的离子电导率,通过添加低熔点的二甲醚醚溶剂来调整其熔点。添加1.3 M LiTFSI(命名为PCE)形成的共溶剂具有22°C的熔点(相变点)和与温度相关的结晶度。DME的引入略微降低了抗氧化电位(4.75 V,图2c),同时显著提高了2个数量级的离子电导率。
如图2b所示,在相变点附近,PCE的离子电导率在20°C时为9.6×10-4 S cm-1,在25°C时为1.11×10-3 S cm-1。斜率在约20°C的拐点处发生显著变化。在较高温度区域(20至45°C),计算得到的PCE活化能为75 kJ mol-1,是较低温度区域(0至20°C)的4倍。这是由于PCE在其熔点以下部分结晶。值得注意的是,较低温度区域的斜率保持不变,这与Arrhenius模型一致,表明PCE中的离子传输是通过溶剂分子的长程运动通过跳跃机制发生的。
相比之下,其中一种组分DME-LiTFSI的温度依赖性离子电导率在σ log和1000 T-1之间呈现线性相关性,而DDCA-LiTFSI表现出三阶段离子电导率转变,这可能与变化有关不同温度下的物理状态。
在锂负极方面,合理设计电解液成分可以抑制锂枝晶和溶剂的过度消耗。据报道,醚-酯混合电解质可稳定锂沉积/剥离并实现稳健的SEI。为了检查PCE和其他样品与锂金属的相容性,使用扫描电子显微镜(SEM)观察沉积在铜箔上的锂的形态(2 mAh cm-2的Li(0.2 mA cm-2 10小时,25°C))。
为了与目前使用的低熔点液态电解质进行更严格的比较,酯基电解质(EC:DMC中的1.0 M LiPF6=1:1 wt%)和醚基电解质(DME:DOL中的1.0 M LiTFSI=1:1 vol%)进行了系统分析和比较。如图2d1所示,在PCE中沉积的锂的形态光滑而致密,而在酯基电解质中则表现出典型的枝晶状形态(图2d2)。
其他样品(SF、SN、DDCA、EC)要么在铜箔上沉积稀缺的锂,要么表现出严重的枝晶状沉积形态。为了进一步说明锂沉积行为,锂形态的演变也可以反映在Li||Li电池的极化曲线中。PCE的电压分布是平坦的(图2e1),表明锂的均匀沉积,这与沉积锂的形态一致。相比之下,酯基电解质在沉积/剥离过程中表现出“峰谷”电压曲线。这种上升和下降现象取决于随时间变化的锂表面形态。在循环过程中,枝晶的重复形成和生长导致电压分布的显著转变和液态电解质中大量的死锂(图2e2)。在可变温度测试条件下(在35°C下沉积和在15°C下剥离),差异是明显的。
尽管这种交替温度测试导致PCE发生相变,而醚和酯电解质始终保持液态,但PCE电池中的沉积锂更加均匀,数量也超过了同类产品。因此,PCE不仅在所选的高熔点溶剂中表现出优异的整体性能,而且在调节锂沉积层方面也优于液态醚和酯基电解质。此外,还进行了不同温度下锂对称电池的阻抗测试。随着温度的降低,界面RSEI从13 Ω增加到256 Ω,显示了PCE固化对离子传导的影响。
总体比较总结在图2f中。PCE在许多方面都优于其他样品,特别是在与Li的相容性和离子电导率方面。 沉积锂的形态通常与离子传导机制和SEI的组成有关。液态PCE中的Li+迁移数(tLi+)为0.58,比酯基电解质中的迁移数(0.17)高3.4倍。PCE在其固态也具有类似的0.53的tLi+。
对于液态PCE,DDCA是一种具有丰富亚甲基的长链分子。-H和TFSI阴离子之间的强静电吸引力阻碍了阴离子的运输。此外,DDCA长链分子的空间位阻和与附近阴离子的纠缠可能使TFSI-更加固定。因此,总体t+远高于传统的液态电解质。这种离子过滤效应导致高tLi+,在Li表面附近产生较弱的空间电荷,因此锂离子分布均匀,锂生长非树枝状。X射线光电子能谱(XPS)也用于研究负极SEI组成在选定的电解质中。在O 1s光谱中,PCE中的SEI表现出C−O(532.6 eV)和C·O/C-OLi(531.8 eV)的峰。
相比之下,P−O的两个新峰(534.1 eV)和P=O/P−OLi(531.2 eV)出现在酯基电解质中。结合F 1s光谱揭示酯基电解质中的LixPOy-1Fz+1基团,可以得出结论,使用PCE可以避免LiPF6的分解(通常会产生HF并腐蚀正极和负极)。PCE中的SEI结合了有机和无机成分,使其具有高稳定性和灵活性。
PCE中的对称电池可以在0.2 mA cm-2下稳定运行超过1200小时,并且在第850次循环时过电位保持在~35 mV,这证明了锂的长期稳定性。 离子传输能力与LSS密切相关。Li+中心的配位环境是通过测量五个样品的拉曼模式变化来确定的,即纯DDCA、LiTFSI、纯DME/DDCA溶剂、液态的PCE(标记为PCE-L)和固态的PCE(标记为PCE-S)。
如图3a所示,在晶体LiTFSI中,位于约755 cm-1处的峰归因于TFSI-的特征拉伸,包括S-N拉伸、C-S拉伸和-CF3弯曲。在PCE-L中,该峰红移至743 cm-1,这意味着TFSI-和Li+之间的相互作用减弱,其中溶剂分子取代了Li+中心周围的TFSI-。
同时,DME/DDCA溶剂的信号峰在PCE-L中从850 cm-1蓝移到872 cm-1,证实了DME/DDCA溶剂分子与Li+之间的配位。在PCE-S中,884 cm-1处的明显峰代表冷冻DDCA分子的散射模型。图3b显示了TFSI-振动模式的更近视图。根据结晶LiTFSI的拉曼光谱对TFSI-的特征光谱进行解卷积。
形成了四种溶剂化物,解离峰位于738、742、747和755 cm-1,它们来自自由阴离子、SSIP、CIP(TFSI-配位到一个Li+阳离子)和聚集体(AGG,TFSI−分别与另外两个Li+阳离子配位)。当PCE从液态转变为固态时,自由阴离子和SSIP配位减少,而CIP配位占更大比例。
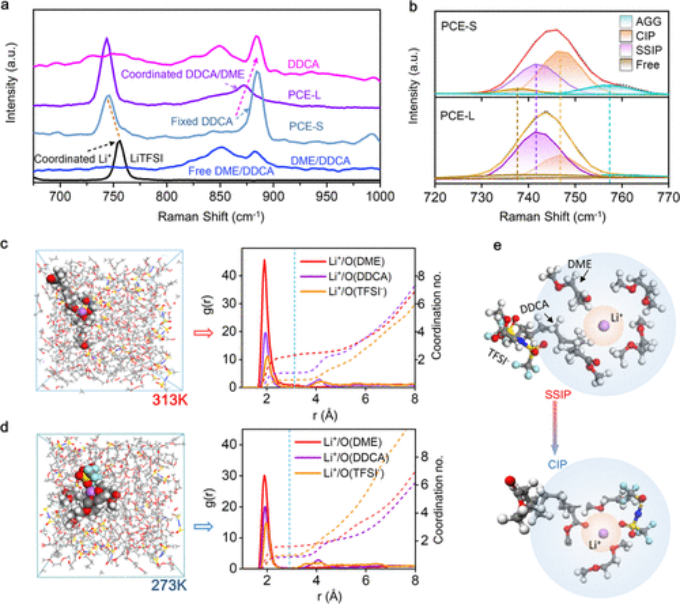
【图3】液态和固态PCE中溶剂化结构的演变。(a)研究样品的拉曼光谱。(b)液相和固相中TFSI-配位的拉曼拟合结果。(c,d)分别从273和313 K的MD模拟获得的快照(左)和RDF和配位数(右)。(X轴表示分离距离,左侧Y轴表示分子的局部密度与中心分子r周围距离处的体积密度的比值)。(e)示意图显示了溶剂化结构从SSIP到CIP的变化。
为了更深入地了解PCE的LSS,应用MD模拟来分析Li+在两种相态中的局部配位。两个温度(313和283K)下的平衡电解质电池轨迹如图3c所示。计算了相应的径向分布函数(RDF)和配位数(Li+第一溶剂化壳,主要由Li+-O对组成),如图3d所示。在313 K的高温下,Li-O(DME)的相互作用强度最强,而Li-O(TFSI-)的相互作用强度最弱。
DDCA、DME和TFSI-中O原子中Li+的配位数分别为2.4、1.2和0.5。该结果符合SSIP溶剂化结构。在283 K的温度下,Li-O(DME)仍然在第一个溶剂化壳层中占主导地位,但Li-O(TFSI-)的贡献增加。Li-O(DME)、Li-O(DDCA)和Li-O(TFSI-)的配位数分别为1.8、0.94和1,对应于高盐浓度电解质中常见的CIP溶剂化结构.拉曼和MD结果表明,LSS经历了从以SSIP为主的液态溶剂化物到以CIP为主的固态溶剂化物的逐渐转变(图3e)。Li+配位环境的转变可以解释为:在固态PCE中,冻结的溶剂分子凝聚了Li盐浓度,形成了局部高浓度的“盐中溶剂”结构。
因此,虽然粘度随着温度的降低而增加,但溶剂化的Li+离子的数量随着大溶剂化壳的解离而减少。此外,即使整个PCE基质变成固态,DME分子仍保持液态。移动的DME可以形成微快离子导电域,从而导致Li+的有效传输,并解释了在上述相变点以下的高离子电导率。
温度引起的相变和PCE的可变形LSS启发利用这些特性来解决正极侧的长期问题。LiMn2O4(LMO)作为具有代表性的TMO正极之一,存在严重的阳离子溶解,在运行过程中往往会导致容量快速衰减。溶解过程主要由LMO正极和液态电解质之间的界面反应引起。因此,抑制接触区域中的Mn(III)歧化是一个明确的解决方案。反应方程式如下所示(eq1),其中n是溶剂化溶剂分子的数量,S是溶剂分子。
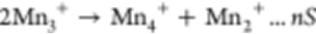
(1)
低于相变点的PCE具有固相和CIP结构,其特点是局部盐浓度高且溶剂不易接近,形成限制金属离子溶解的天然物理屏障(图4a)。为了验证这一点,LMO正极与PCE配对,而商业酯基电解质作为对照样品。通过电感耦合等离子体发射光谱法(ICP-OES)分析LMO-Li电池充电/放电过程后的隔膜,对Mn离子溶解进行了定量分析。
在图4b中,在3.3 V/15°C放电特定时间后,PCE内检测到的Mn离子浓度为酯基电解质的1/3。之后,将放电的正极拆开并重新组装以测试剩余容量和库仑效率(CE)。如图4c所示,PCE中的剩余容量和CE为105 mAh g-1,低温放电后为93%。相比之下,酯基电解质中的相应值仅为85.4 mAh g-1和90.5%。
据报道,在充电和放电过程中都会发生溶解;然而,结果表明,在放电过程中,Mn离子的损失非常严重。PCE中的锂和隔膜比其他电解质中的更亮、更清洁。锂和隔膜上的黑色和棕色物质可能与酯和醚电解质中的分解和Mn离子溶解有关。
因此,通过固化,在放电过程中应保持更多的活性物质。还评估了在35°C充电后的Mn离子溶解。在此温度下,酯基电解质甚至促进了溶解,因为LiPF6与残留的水反应形成HF。然后在PCE-S中放电的LMO正极上进行X射线衍射(XRD)图案。如图4d所示,收集的正极材料显示出主要的LiMn2O4特征峰,证实了空间群为Fd3̅m的立方尖晶石结构不变。
对于酯基电解质中的正极材料,在32.9°、38.8°和45.1°处出现了新的显著峰,表明与大量Mn离子溶解相关的结构损坏。此外,在Cu/Li负极表面上进行了XPS循环30次后(图4e)。Mn的信号峰仅存在于酯基电解质的电池中,表明Mn离子溶解行为不受限制。上述特性直接证明PCE-S物理阻断了Mn离子的溶解并保持了LMO的结构完整性正极。
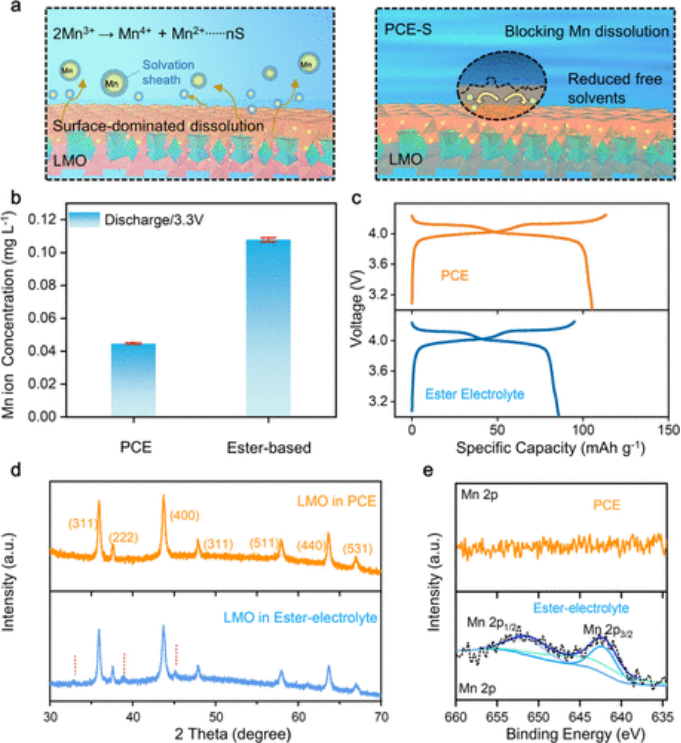
【图4】阻止正极锰离子溶解在PCE中。(a)显示酯基电解质(左)和PCE-S(右)之间不同Mn离子溶解行为的示意图。(b)ICP-OES结果显示PCE-S和酯基电解质中的Mn离子浓度。(c)充电/放电曲线显示PCE-S和酯基电解质中的容量保持率和库仑效率(CE)。(d)XRD图案显示LMO结构在20个循环后发生变化。(e)PCE和酯基电解质中负极的XPS分析。
由于PCE对正极和锂负极都有利,为了放大LMO的溶解现象并测试阻断效果,组装好的Li-LMO电池首先在苛刻的交替温度条件下进行循环(第一次循环是通过恒定-电压在4.25 V/35°C下充电17小时,在15°C下放电13小时,然后进行10个正常循环(3.3-4.25 V))。
酯基电解质中的电池表现出较低的初始容量(16.18 mAh g-1,尽管在第二次循环时恢复到88.6)和快速的容量衰减(从第2次到第11个循环)和第二个充电过程经历了剧烈的波动,表明Mn离子的穿梭效应。醚基电解质中的电池表现出更差的性能(从42 mAh g-1的初始容量到第11次循环时几乎为零容量),这可能是由于醚电解质在高电压和高温下的分解。
相比之下,PCE中电池的充电/放电曲线正常,容量保持率最高(从128到64 mAh g-1)。在交替温度测试下,Mn离子的溶解加速,这直接反映在商用酯基和醚基电解质中电池性能的恶化。
然而,由于在溶解较严重的放电状态下物理阻塞效应和游离溶剂减少,PCE可以抑制Mn的溶解,保护LMO结构不被破坏。 正如所证明的,在放电过程中,Mn离子损失更为严重。为了利用这个问题并提高LMO正极的容量保持率,通过采用一种新颖的交替温度循环协议,利用了相变特性的优势(图5a)。
具体来说,在一个典型的循环中,使用PCE作为电解质的电池首先在35°C(PCE-S)下充电(0.2 C),直到达到高截止电压。然后,当温度降至15°C(PCE-S)时,电池会静置特定时间以达到热力学平衡。最后,电池在0.1 C下放电,直到达到低截止电压,完成一个完整的循环。这种交替温度协议将PCE-S在15°C时的离子电导率、锂离子沉积和能耗考虑在内。
温度过低或过高都会导致不必要的容量损失和能源浪费。为了比较,电池也在35或15°C的恒定温度下循环。如图5b所示,交替循环电池在第20次循环时表现出121 mAh g-1的高比容量,优于初始容量最高的35°C循环电池。在120次循环后,使用交替循环协议的电池保持98 mAh g-1的比容量,而35°C和15°C循环的电池的比容量分别仅为34和78 mAh g-1。
交替循环、15°C循环和35°C循环电池的CE分别为98.5%、99.3%和68.5%。因此,交替循环协议使PCE在阻止Mn离子溶解(在放电过程中)和调节锂沉积(在充电过程中)方面具有优势,实现了长期循环稳定性。此外,还测试了具有不同DDCA/DME比例的LMO-Li电池的电化学性能。
同样,优化的PCE(DDCA/DME=3)表现出最佳的容量保持率和稳定性。LMO-Li电池的倍率性能和PCE与LiNi0.6Mn0.2Co0.2O2(NMC)正极的应用证明了PCE对先进正极的普遍适用性。
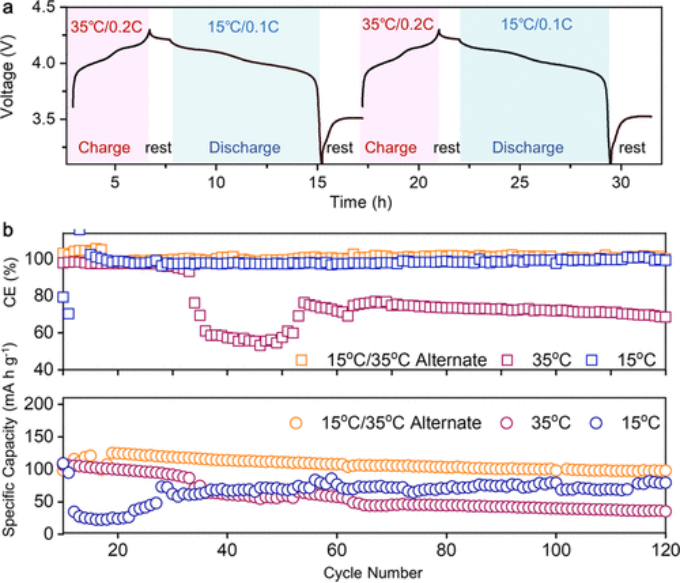
【图5】Li-LMO电池在交替温度条件下的电化学性能。(a)非典型电压曲线显示温度交替循环协议。(b)LMO-Li电池在PCE中的库仑效率(上)和循环性能(下),分别使用交替循环、35°C循环和15°C循环协议。
四、总结与展望
通过简单地调节由LiTFSI锂盐和DDCA和DME的混合助溶剂组成的PCE的外部温度和相态,实现了LSS的内调节。通过实验和计算研究了其物理特性和电化学性能。发现LSS可以根据PCE的物理状态在SSIP和CIP之间转换。LSS的原位转变使PCE即使在其固态下也具有高离子电导率。
此外,与广泛使用的液态电解质(酯和醚电解质)相比,PCE在调节锂离子沉积和抑制锰离子在LMO正极中溶解方面表现出优越性,尤其是在不同温度下。基于PCE的独特性质,提出了一种交替温度循环协议(15°C充电/35°C放电),并且Li-LMO电池的性能优于传统的液态酯和醚电解质。电解液LSS的工作调整策略将启发未来智能电解液的设计。
审核编辑:刘清
-
我想自己测试电解质2013-03-09 2807
-
超薄电解质电容器问世 手机可迎袖珍化时代2014-09-24 3353
-
电池内的电解质是什么?2009-10-20 1237
-
超晶格电解质材料2009-11-10 971
-
美国开发出一种新型阴极和电解质系统 有望改善锂离子电池2019-09-16 1644
-
电池电解液和电解质的区别_电池电解液和电解质的两种形态2020-04-16 25737
-
宁德时代公开“一种固态电解质的制备方法”专利2021-01-20 4182
-
浓度极化诱导相变稳定聚合物电解质中的锂镀2022-09-06 2970
-
钠离子电池的电解质分类2022-10-09 6740
-
相变电解质助力高稳定性锂金属电池2022-10-25 3389
-
开发一种不可燃的液态聚合物电解质2022-12-05 2330
-
上海电力大学《AFM》:一种新型复合固态电解质设计!2023-02-06 2022
-
一种新型的ZnSO4-基共晶电解质介绍2023-08-14 3866
-
高锂金属负极形貌稳定性的聚电解质2023-08-16 2447
-
无极电容器有电解质吗,无极电容器电解质怎么测2024-10-01 2063
全部0条评论

快来发表一下你的评论吧 !
