

应变效应对催化剂活性的影响
描述
0 1 引言
单原子催化剂(SACs)结合了均相催化剂的高活性和非均相催化剂的稳定性,在各种反应中都有很大的潜力,包括析氢(HERs)和析氧(OERs)反应。为SACs选择合适的衬底是很重要的,它可以有效地固定单个原子,防止其团聚,同时可以提供丰富的电子。作为一种特殊的三元过渡金属硫化物,“Janus” MoSSe的带隙和层间距介于在MoS2和MoSe2之间,对水分子的吸附能力也要强于MoS2和MoSe2。结构对称性的破坏导致了体系电子的重新分配,并有效地积累电荷到相应一侧的Janus单分子层金属上。因此,在“Janus”MoSSe表面负载合适的过渡金属有望构建高效、稳定的全分解水电催化剂。
0 2 成果简介
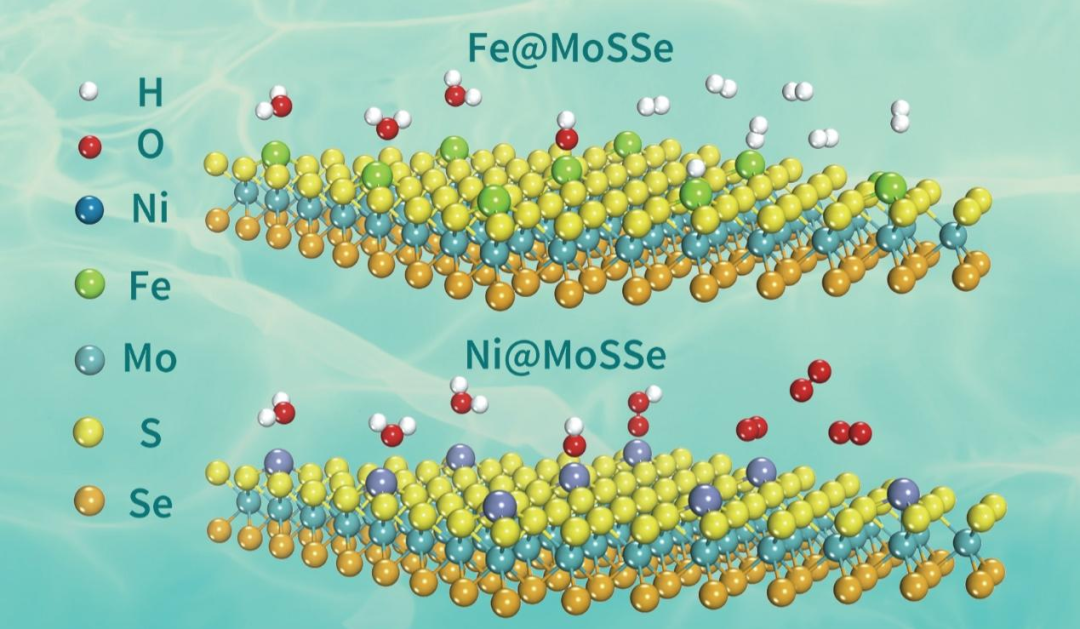
图1 催化剂筛选结果示意图
该工作利用密度泛函理论(DFT)计算在一系列的单原子过渡金属锚定的Janus MoSSe催化剂中,筛选出稳定、高效的析氢(HERs)和析氧(OERs)反应电催化剂。与以往的工作不同之处在于,利用鸿之微DS-PAW程序对催化剂进行AIMD模拟,从动力学和热力学双角度来判断所设计催化剂的稳定性。最终筛选结果表明,Fe@MoSSe具有出优异的HERs活性,由于低廉的价格使得其更具有实际应用的潜力。同时Ni@MoSSe表现出极低的OER过电位(0.32 V)和优异的OERs催化性能,也是一种有望实际应用的电催化剂。基于筛选结果,进一步深入研究了HERs和OERs的催化性能和活性来源,并进一步探讨了应变效应对催化剂活性的影响。
0 3 图文导读 该工作将一系列的过渡金属单原子锚定在MoSSe的表面,构建出TM@MoSSe催化剂。为筛选出不同TM@MoSSe催化剂催化稳定的结构,首先计算了过渡金属单原子在表面不同位点处的吸附能,确定最稳定的吸附位点为Mo位点。随后,比较催化剂的溶解势,进一步确定材料在电化学反应环境中的稳定性。溶解势的计算结果与吸附能一致,即TM@MoSSe的稳定性模型是过渡金属单原子锚定在MoSSe表面的Mo原子位点。单靠热动力学计算不足以描述材料在实际应用温度下的稳定性。在能量计算的基础上,通过AIMD计算进一步确定材料的稳定性。结果筛选出单一的Fe、Co、Ni、Ru、Rh、Pd、Ir、Pt单原子锚定在MoSSe表面是可以构成稳定的催化剂。
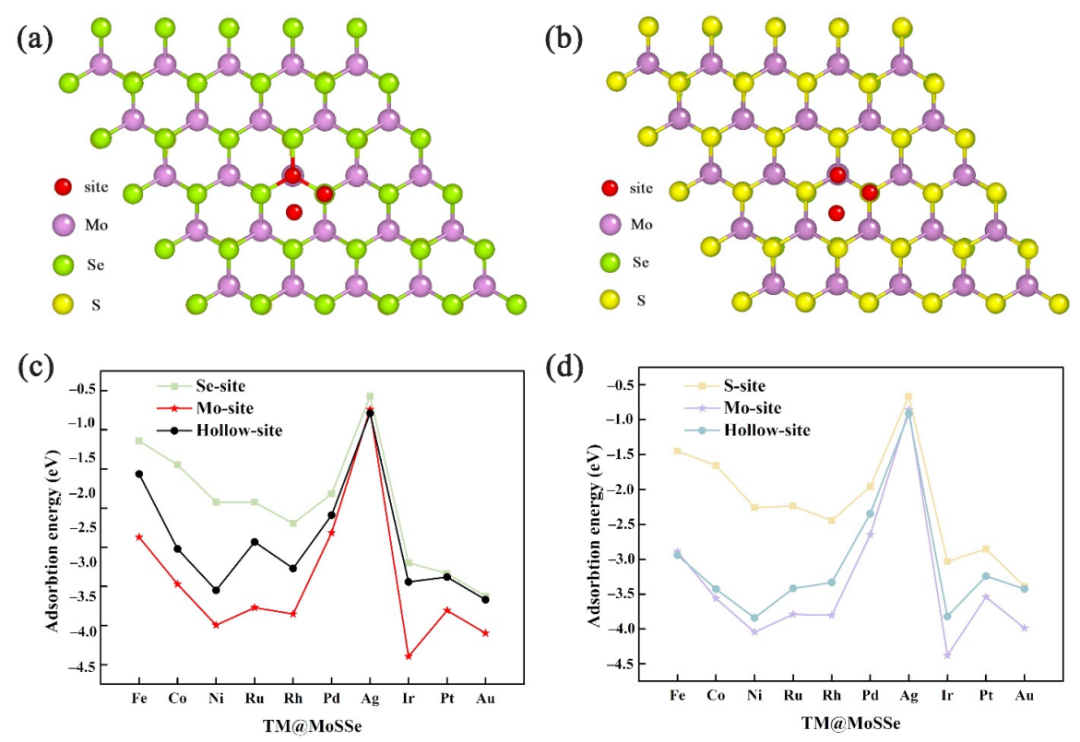
图2 过渡金属原子的吸附结构固定在(a) MoSSe的S侧和(b) MoSSe的Se侧的示意图。不同过渡金属原子在(c) MoSSe的Se侧和(d) MoSS的S侧的吸附能
为了评价TM@MoSSe的HER和OER活性,基于Norskov教授提出的计算氢电极方法计算了所有TM@MoSSe催化剂上HER和OER每一步反应的吉布斯自由能,并确定过电位。结果表明Fe@MoSSe、Ru@MoSSe、Rh@MoSSe和Pt@MoSSe均具有较低的析氢反应过电位,结合实际应用的经济成本问题,Fe@MoSSe可以作为一种潜在的HER电催化材料。,在一系列的TM@MoSSe中,Ni@MoSSe的OER过电位值最小(η = 0.32 V),OH*解离生成O*是OER的决速步骤。
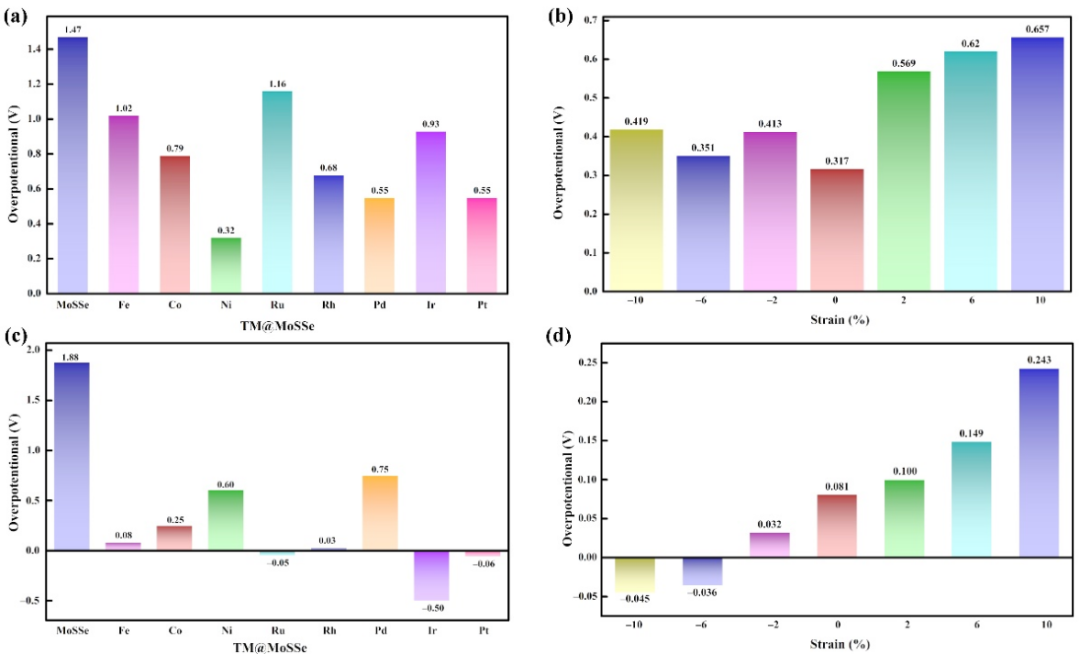
图3(a) TM@MoSSe和(b) Ni@MoSSe在不同应变下的OER过电位; (c) TM@MoSSe和(d) Fe@MoSSe在不同应变下的HER过电位。
过渡金属与MoSSe组合成的单原子催化剂具有优异的催化性能,为了理清优良催化活性的来源,计算了TM@MoSSe催化剂的电子结构信息。结果分析发现,过渡金属锚定在MoSSe表面后,将体系的电荷重新分配,并极大的提高了催化剂的导电性,进一步增强催化活性。发现催化剂的d带中心与OER过电位之间存在良好的线性关系,在TM@MoSSe体系中,随着过渡金属与MoSSe表面结合强度的增加,催化剂的稳定性和活性得到提高,材料的OER过电位降低。最后考察了应变效应的影响对催化活性的影响。将态密度计算结果与催化剂的d-band中心联系起来,发现拉伸或者压缩应变改变了二维材料表面原子的重叠度,dx2、dxy和dz2轨道之间的相互作用对材料体系中d-band中心位置的变化起着重要作用。
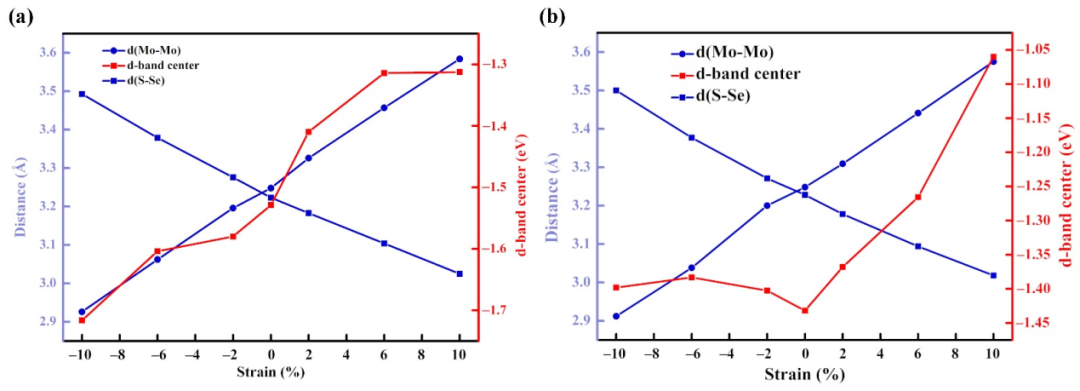
图四 应变效应对(a) Ni@MoSSe和(b) Fe@MoSSe的d带中心的影响
0 4 小结
这项工作基于密度泛函理论计算,筛选出稳定、高效具有应用价值的析氢和析氧反应过渡金属MoSSe单原子催化剂。同时,强调单纯从热力学结果来判断材料的稳定性是不够充分的,需要结合动力学模拟进一步判断。深入研究催化活性来源以及应变效应对催化活性的影响,为实验设计、制备高效稳定的分解水催化剂提供直接的理论指导。
-
网关助力催化剂产业升级,解决痛点问题!2024-08-14 1133
-
构建提高酸性水氧化催化剂稳定性的氧扩散路径2023-08-02 2123
-
催化剂分离和循环利用问题2023-05-23 2061
-
如何提高HEAs催化剂的催化活性和优选设计研究2022-12-14 1992
-
“纳米岛”型催化剂突破传统催化剂活性和稳定性的矛盾2022-11-18 1893
-
揭示尖晶石催化剂几何构型影响HER活性的机理2022-11-03 5808
-
Mo配位FeCoNiMo碳负载高熵电催化析氧催化剂图文解析2022-09-20 3613
-
一类新的钌基催化剂,采用原位制备技术2022-08-13 3177
-
低结晶和异质结构AuPt-Ru@CNT像高效多功能电催化剂2022-05-31 810
-
高活性生物质碳负载Fe/Pt单原子双功能催化剂开发2021-02-12 3725
-
纳米笼催化剂保障燃料电池超长稳定运行时间2019-11-21 3732
-
碱性醇类燃料电池新型催化剂的研究2011-03-11 2003
-
燃料电池氧电极催化剂的研究2011-02-22 773
-
脱氧催化剂纯化氢气的应用研究2009-01-10 1222
全部0条评论

快来发表一下你的评论吧 !
