

利用电分析和纳米尺度表征方法证明锂电镀形貌和电流密度高度相关
电池
描述
研究背景
安全耐用的锂金属电池需要均匀的锂沉积形貌。电解液修饰能够调控锂沉积,并提高电池的可循环性。然而,电池在快充条件下还是能够诱导枝晶状锂沉积,降低循环稳定性。
因此,解析Li沉积路径,并确定快充过程中Li枝晶的起源是开发快充锂金属电池的关键。
成果简介
近日,斯坦福大学崔屹&鲍哲南教授在Nano Letters上发表了题为“Resolving Current-Dependent Regimes of Electroplating Mechanisms for Fast Charging Lithium Metal Anodes”的论文。该论文开发了一种结合电分析和纳米尺度表征的方法来证明锂电镀机制和形貌与电流密度高度相关。
对Li+通过固体电解质界面膜(SEI)输运的测量表明,低电流可在Li||SEI界面处引发锂电镀,而高电流会引发SEI破裂和在Li||电解质界面处锂的电镀。后一种途径可以在极高的电流下诱导锂沿{110}面均匀生长。SEI破裂会产生有害的形貌和较差的可循环性。因此,避免SEI破裂和电解质中缓慢的离子运输是必不可少的。
研究亮点
(1)本工作利用电分析和纳米尺度表征方法的互补优势,证明了锂电镀形貌和电流密度高度相关。
(2)首次采用低温透射电镜(cryo-TEM)报道了SEI的Li+输运特性,并建立了三种与电流相关的电镀机制。
(3)结果表明,SEI破裂是电池快充期间形成锂枝晶的主要原因。
图文导读
描述Li+在SEI中的传输参数,如扩散系数(DLi+SEI)、离子电导率(λ)和载流子浓度(n0),对于区分电镀机制是至关重要的。本工作将Li||Li对称电池的EIS数据和低温TEM测量的原生SEI厚度相结合,来计算这些参数(图1a-d)。一般而言,98%的界面阻抗来自于Li+通过SEI的输运。从EIS计算传输参数需要可靠的SEI厚度和等效电路模型。
电镀锂的玻璃化及其SEI和低温TEM成像可以保持原始SEI厚度。 图1b 的TEM图像显示,SEI厚度约为20 nm。锂与LiPF6碳酸乙烯:碳酸二乙酯(EC:DEC,体积比1:1)电解质之间的阻抗与通过Li2O SEI模型量化锂离子输运的等效电路非常吻合(图1a)。界面电阻约为150 Ω·cm2,计算出DLi+SEI=2.2×10-9 cm2/s,λ=1.6×10-8 s /cm, n0=2.1×10-6 mol/cm3。
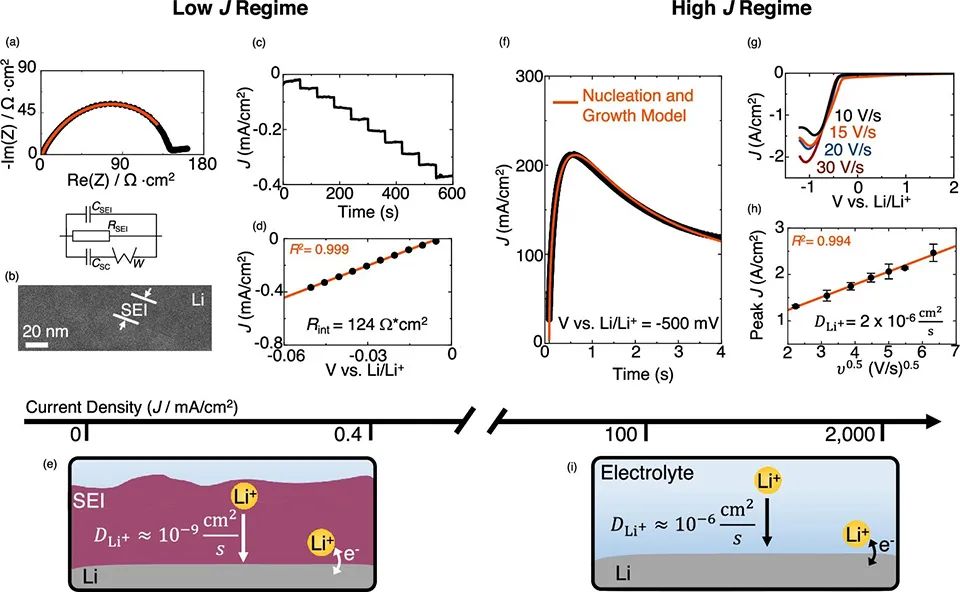
图 1、(a)Li||Li电池的典型Nyquist图和等效电路。(b)电解质中玻璃化锂的低温高分辨率TEM图像,其显示了锂表面SEI的厚度。(c)用阶梯伏安法在三电极电池中镀锂的电流分布。(d)来自(c)图的稳态线性J-E关系。(e)低充电速率下的锂电镀路径示意图。(f)−500 mV电压阶跃下,锂电镀的电流分布。(g)钨UME上镀Li的LSV曲线显示了峰J和E之间的关系。(h)(g)图中峰电流随v1/2的变化。(i)极快充电速率下的电镀路径示意图。
为了测试离子通过SEI的传输是否在更动态的条件下受到速率限制,采用三电极阶梯伏安法将锂电镀到锂电极上(图1c)。在从−5 mV到−50 mV的范围内,可以观察到连续阶跃的稳态电流密度(J)曲线。这表明液体电解质中不存在浓度梯度,因为当存在Li+浓度梯度时,J曲线会随时间衰减。J-E图也是线性的,电阻约为124 Ω·cm2(图1d)。该值与EIS结果一致,表明当J小于约0.4 mA/cm2时,电镀发生在Li||SEI界面(图1e)。
图a-d显示,在低J下,电荷转移发生在Li||SEI界面,但一些研究表明,在高J时,电镀发生在Li||电解质界面。这些报道称,用超微米电极(UME)沉积Li的J高达2000 mA/cm2,此时Li电镀速度超过SEI形成速度。当SEI面电阻为100 Ω·cm2时,在2000 mA/cm2下沉积锂需要200 V,这有可能在Li||电解质界面处破裂SEI,并引发新的锂电镀。
为了验证这一假设,采用瞬态线性扫描伏安法(LSV)和大的电压(E)阶跃,即用100-2000 mA/cm2的J电镀锂(图1f-i)。 图1g显示,在不同扫速(v)下,克服成核势垒后,J随Li金属的生长而急剧增加,直至J达到扩散限制峰(Jp)。图1h显示了不可逆电极反应的v1/2和Jp之间的线性关系。有效扩散系数(DLi+)可以用
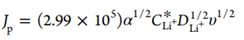
其中α为传递系数,CLi+*为Li+的体积浓度。假设α为0.5,算出来的DLi+为2×10 -6 cm2/s,这与LiPF6 in EC:DEC中的DLi+一致,且比DLi+SEI大3个数量级,表明SEI没有阻碍锂的电镀,并且在锂||电解质界面发生了界面电荷转移(图1i)。
为了进一步确定在如此高的J下,Li||电解液界面上是否会发生Li电镀,使用大E阶跃法,在J>100 mA/cm2下电镀Li。图1f显示,在−500 mV下,J曲线很好地符合非活性金属形核和生长的模型。拟合得出的DLi+值也比DLi+SEI高3-4个数量级。上述结果表明,在如此高的J下,新的Li电镀发生在Li||电解质界面(图1i)。
为了评估在锂||电解质界面和高J下的电镀如何影响锂的形貌,用扫描电子显微镜(SEM)对高J(1000 mA/cm2)下在UME上电镀的锂进行了成像。当J为10 mA/cm2时,锂沉积形貌为丝状(图2d);然而,当J为100 mA/cm2时,颗粒状锂出现(图2c),超过1000 mA/cm2时,锂仅以菱形十二面体的形式生长(图2a,b)。图2B中突出显示的菱形十二面体是{110}面体心立方(BCC)晶体的特征:{110}面是像Li这样的BCC金属中最常见的堆积平面。

图 2、在1000 mA/cm2(a,b)、100 mA/cm2(c)和10 mA/cm2(d)下,钨UME上电沉积的Li金属微观结构。(b)扫描电镜图像显示菱形十二面体的Li具有清晰的{110}面。(e)在100 mA/cm2下电沉积的Li冷冻TEM图像。(f)(e)中粒子的SAED图;(g)位于(e)红框内Li(110)表面和SEI之间的界面的低温高分辨率TEM图像。
图2e显示,沿着〈111〉轴排列的Li颗粒为BCC Li晶体,具有六边形SAED图案(图2f)。SAED图案对应于{110}晶面族。图2g显示,Li(110)和SEI之间具有良好的原子级界面。
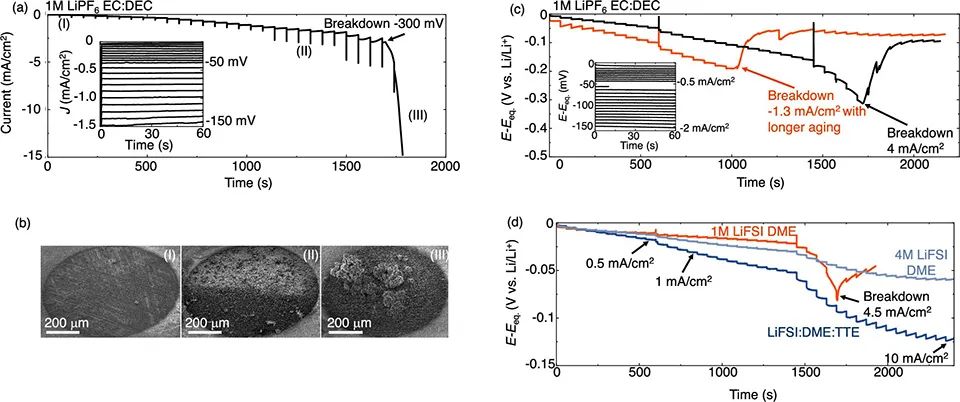
图 3、(a)LiPF6 in EC:DEC中锂电沉积的阶梯伏安法曲线。(b)在(a)中阶梯伏安法测量的不同阶段Li的形貌。(c)LiPF6/EC:DEC中锂电沉积的阶梯恒流法电压曲线。(d)不同电解质中的Jcrit.SEI。
在图3a的低J区(I),非原位扫描电镜显示,Li的形貌较为均匀(图3b (I))。在图3a的区域(II)中,Li+开始在电解质中扩散,Li的电镀基本保持均匀(图3b (II))。SEI在区域(III)分解后,高表面积的Li枝晶清晰可见(图3b (III))。这些图像表明,在电池快充期间,SEI破裂引发了高表面积的“枝晶状”锂。
图3c显示了从0.05 mA/cm2到8 mA/cm2恒流阶跃下的电压曲线。对于低J,电压曲线平坦。一旦J达到1.5 mA/cm2(约−120 mV),电压曲线偏离欧姆行为,并在单个J阶跃中随时间增加。在4.5 mA/cm2(约−300 mV)下,E迅速降低,表明SEI破裂,并暴露出Li表面。
因此,将J=4.5 mA/cm2定义为SEI(Jcrit.SEI)破裂的临界电流密度。Jcrit.SEI也取决于电解质。图3d显示,标准醚电解质(1 M LiFSI in DME)中的Jcrit.SEI也为4.5 mA/cm2。然而,在最先进的电解质(4 M LiFSI in DME和LiFSITTE)中,SEI破裂现象不明显。
图4a, b显示,1 M和4 M LiFSI电解质在1~20循环中均有稳定的库伦效率,分别为95.8%±0.5和96.3%±0.7(1 mA/cm2, 1 mAh/cm2)。然而,在5 mA/cm2沉积时,只有4 M LiFSI电解质保持稳定的库伦效率(93.6%±0.9)。脉冲充电也可能会阻止SEI破裂,以及Li枝晶的生长(图4c)。
在短脉冲的慢速和快速充电模式之间切换,可以通过在低电流密度脉冲期间修复断裂的SEI来防止枝晶传播。图4d-f显示,在1.5C和0.05C之间切换的脉冲充电模式延长了快充锂金属电池的寿命。
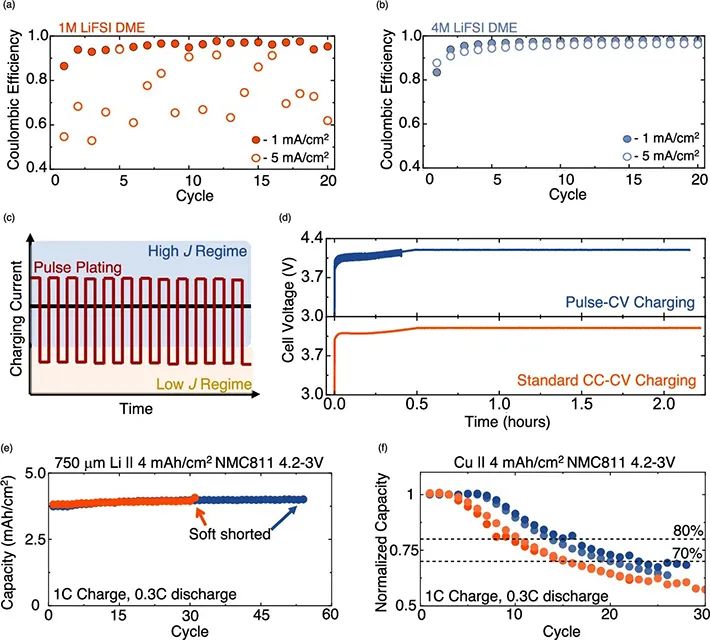
图 4、(a)1 M LiFSI DME和(b)4 M LiFSI DME中,不同充电速率下锂沉积的库伦效率。(c)在高J和低J之间切换的脉冲充电模式示意图。(d)Li||NMC811电池在脉冲CV和标准CCCV充电模式下的电压曲线。(e)Li||NMC811电池的循环性能。(f)无负极Li||NMC811电池的循环性能。
总结与展望
本工作将锂金属负极的充电速率、电镀机理和形貌联系起来。通过测量SEI中Li+的输运,可以将Li电镀分为三种状态(图5)。缓慢的电镀状态对应于大约1 mA/cm2以下的电流。此时,电镀发生在Li||SEI界面,Li+通过SEI的输运速率是有限的。
在这种状态下,锂的形貌更加均匀。电流超过100 mA/cm2对应于极快的电镀速率。此时,电镀破坏了SEI,并发生在新沉积Li||电解质界面上。Li可以均匀生长为{110}面菱形十二面体,新鲜的Li表面暴露在电解液中,在较长充电时间后会引发枝晶生长。
在标准碳酸盐电解质中,SEI在约2-4 mA/cm2下破裂,在Li||电解质界面开始发生锂的电镀。破裂引发锂枝晶生长和大量的活性锂损失。电解质的离子导电性差也会导致枝晶生长,但SEI破裂则呈现出一种更为极端的失效模式。因此,除了提高电解质的离子导电性外,缓解SEI的破裂对快充至关重要。
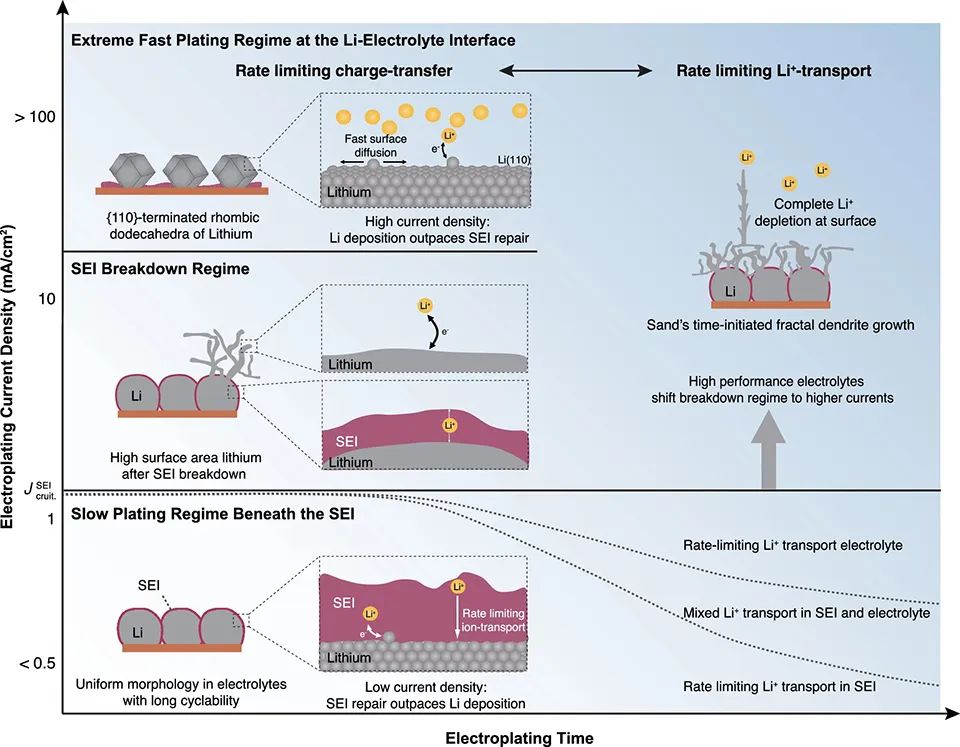
图 5、锂离子形貌随电流的变化和电镀机理。
审核编辑:刘清
-
新型椭圆偏振法SHEL在纳米尺度面积表面测量的应用2025-11-24 2964
-
聚焦离子束技术之纳米尺度2025-04-08 931
-
磁化电流密度和传导电流密度的关系2024-10-09 4383
-
MXene水溶液润滑的长寿命高电流密度摩擦伏特纳米发电机2023-12-11 3259
-
纳米级测量仪器:窥探微观世界的利器2023-10-11 21134
-
什么是电流密度?电流密度过高带来的问题有哪些?2023-09-28 23263
-
电流密度有哪几种形式2021-10-09 3844
-
电流密度分析和电热协同分析2020-07-07 2462
-
纳米尺度下的光和物质强耦合系统的研究2020-04-21 5588
-
硅藻—纳米尺度下天然合成的AFM成像2019-10-28 2210
-
PCB图形电镀夹膜原因分析2018-09-20 6967
-
超全面锂电材料常用表征技术及经典应用举例2016-12-30 4385
-
美国首次展示纳米尺度波导2011-06-04 1227
-
纳米晶PZT薄膜叠层型体声波滤波器的研究2010-04-24 4227
全部0条评论

快来发表一下你的评论吧 !
