

揭示尖晶石催化剂几何构型影响HER活性的机理
描述
研究背景
过渡金属基催化剂的局部几何构型会显著影响其催化活性。为了有效选择和精确构建有利于催化反应的几何构型,阐明不同几何构型对催化活性的具体贡献,并进一步挖掘导致催化活性差异的潜在机制,具有至关重要的研究意义。尖晶石(AB2X4)是一种典型的多配位构型催化剂,其晶体结构中含有四面体配位(A2+−X)Td和八面体配位(B3+−X)Oh,被广泛应用于电催化领域。
然而关于尖晶石催化剂中起主导作用的活性配位构型,仍存在争议。目前,相当多的工作仅仅关注配位环境变化对中心金属位活性的影响,而对配位非金属原子的研究较少。为了精确识别尖晶石催化剂中的不同活性中心,并深入研究HER活性与几何构型之间的关系,碱性HER是一个理想的模型反应。
成果简介
鉴于此,北京科技大学康卓教授、张跃院士团队(共同通讯作者)等通过在Co3O4中掺杂无催化活性的Zn2+和Al3+,分别取代的CoTd2+和Cooh3+。制备的ZnCo2O4和CoAl2O4仅暴露Cooh3+或CoTd2+,以构建(Co3+-O)Oh或(Co2+-O)Td的孤立几何构型,量化了不同配位构型对碱性HER催化活性的贡献差异,阐明了配位中心金属位点与非金属配体位点对水分子解离的协同促进效应。
研究亮点
1、通过取代策略,选择性暴露Co3O4中的单一配位构型,确认了(Co3+-O)Oh构型对Co3O4的HER活性起主导作用;
2、CoOh3+的d带中心上移有利于水分子解离,相应氧配体的缺电子态有利于氢气脱附,从而降低了整个水还原过程中的能垒。
图文介绍
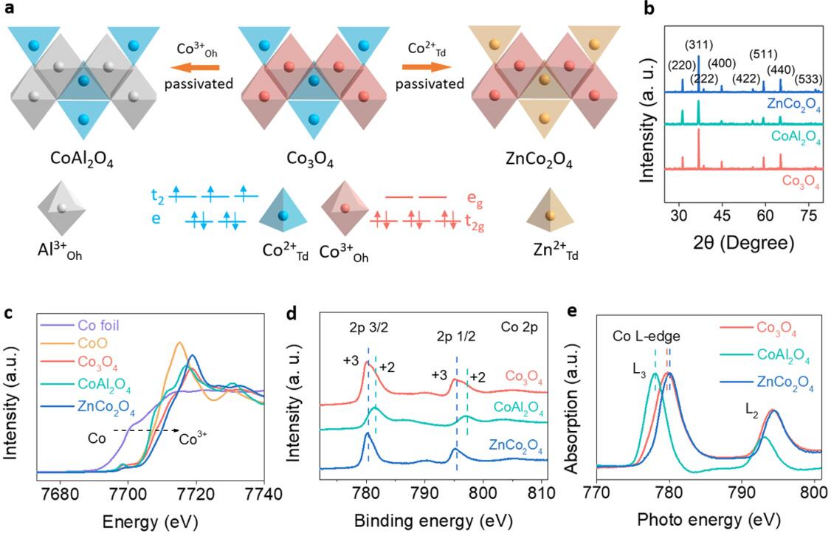
图1 a)CoAl2O4, Co3O4和ZnCo2O4结构示意图;b)XRD图谱;c)归一化非原位Co K边XANES光谱,由此可确定Co3O4、CoAl2O4和ZnCo2O4中Co价态分别为+2,+2~+3和+3价;d)XPS图谱;e)Co L边XANES光谱。
作者研究了尖晶石型Co3O4中(Co2+-O)Td和(Co3+-O)oh的碱性HER活性,分别用无活性的Zn2+和Al3+替代四面体和八面体中的Co2+和Co3+(图1a)。如图1b的XRD图所示,位于2θ=31°、37°、39°、45°、57°、59°和65°处的特征峰归属于立方尖晶石相Co3O4、ZnCo2O4和CoAl2O4的(220)、(311)、(222)、(400)、(422)、(511)和(440)晶面。
通过边缘X射线吸收谱研究了Co3O4、CoAl2O4和ZnCo2O4中Co的价态,如图1c所示。Co价态遵循ZnCo2O4>Co3O4>CoAl2O4的顺序,这一趋势证明了ZnCo2O4和CoAl2O4分别含有Co3+和Co2+。采用XPS光谱(图1d)和XANES光谱(图1e)表征了Co3O4、CoAl2O4和ZnCo2O4的电子结构。
图1d中Co 2p3/2和Co 2p1/2峰来自Co 2p的自旋轨道双重峰。位于781.3和796.81 eV处的两个峰对应于Co2+,而位于780.1和794.85 eV处的峰对应于Co3+,因此,可以确认在CoAl2O4和ZnCo2O4中暴露的Co的价态为+2和+3,而Co2+和Co3+共存于Co3O4中。
ZnCo2O4比CoAl2O4更高的Co价态可归因于ZnCo2O4中Co3+与O-阴离子之间更强的相互作用。图1e展示了Co L边光谱。每个光谱包含对应于较高能量L2和较低能量L3的两个峰,这可以归因于2p的自旋轨道分裂。Co L3,2边XANES光谱中的白线峰与受激电子从Co 2p3/2和Co 2p1/2到未占据的Co 3d态的跃迁相一致。
与Co3O4相比,ZnCo2O4和CoAl2O4的Co L边光谱分别发生了正移和负移,这进一步表明Co3+和O之间的轨道重叠程度比Co2+和O之间的轨道重叠程度更强。
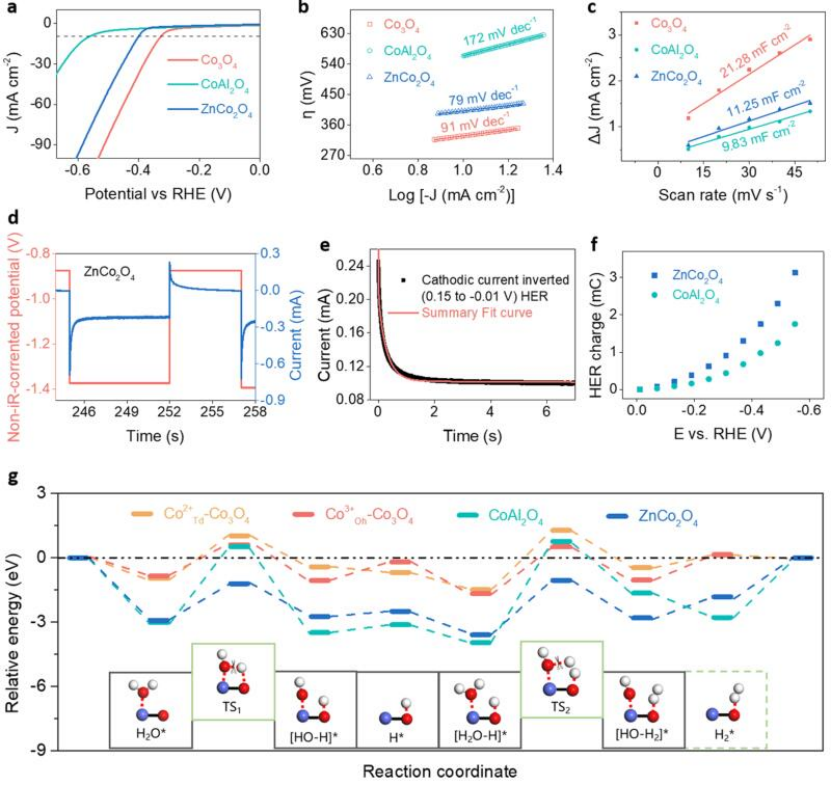
图2 a)HER极化曲线;b)塔菲尔曲线;c)电容电流与扫描速率的线性拟合,其斜率值为Cdl;d)HER脉冲电压及其脉冲电流响应;e)瞬态电流衰减拟合结果;f)阴极电位对应的HER电荷;g)Co2+Td-Co3O4,Co3+Oh-Co3O4,CoAl2O4和ZnCo2O4表面示意图及其在碱性HER过程中的能量变化。
在1.0 M KOH电解质中研究了Co3O4、CoAl2O4和ZnCo2O4的电催化HER活性。如图2a所示,Co3O4、CoAl2O4和ZnCo2O4在10 mA cm-2时的过电位(η10)分别为328、563和401 mV,这表明ZnCo2O4中的(Co3+-O)Oh对碱性HER的催化活性优于CoAl2O4中的(Co2+-O)Td。Co3O4、CoAl2O4和ZnCo2O4的Tafel斜率分别为91、172和79 mV dec-1(图2b)。
ZnCo2O4较低的Tafel斜率表明,与(Co2+-O)Td相比,(Co3+-O)Oh更有利于水分子解离过程,具有更快的反应动力学。如图2c所示,Co3O4样品的Cdl为21.28 mF cm-2,而ZnCo2O4和CoAl2O4的Cdl分别为11.25和9.83 mF cm-2,这表明ZnCo2O4和CoAl2O4具有几乎相同的ECSA,因此这些催化剂之间的HER活性差异不是由ECSA引起的,而是源于不同几何构型之间的本征活性差异。因此,可以初步确定(Co3+-O)Oh构型,为碱性HER提供比(Co2+-O)Td更合适的平台。
如图2d所示,采用PV技术量化了在给定电势下参与法拉第反应的电荷。图2f示出了CoAl2O4和ZnCo2O4的HER电荷随电势的变化。图2e为拟合结果,该结果在标准偏差范围内。与CoAl2O4相比,ZnCo2O4表面有更多的电荷参与法拉第反应,进一步揭示了八面体(Co3+-O)Oh具有更高的HER活性,是Co3O4中主要的活性几何构型。
为了研究上述两种构型催化碱性HER的热力学,作者计算了HER反应过程中的相对能量变化(图2g)。吸附态水分子需要经历一个过渡态(TS1)才能发生HO-H键的断裂。相对于前一状态,Cotd2+-Co3O4、Cooh3+-Co3O4、CoAl2O4 (Cotd2+)和ZnCo2O4 (Cooh3+)的能垒分别为1.98、1.45、3.53和1.71 eV。很明显,ZnCo2O4中Cooh3+处的水分子解离势垒远低于CoAl2O4中Cotd2+处的水分子解离势垒。这表明与Cotd2+相比,吸附在Cooh3+处的水分子解离过程在热力学上更有利。
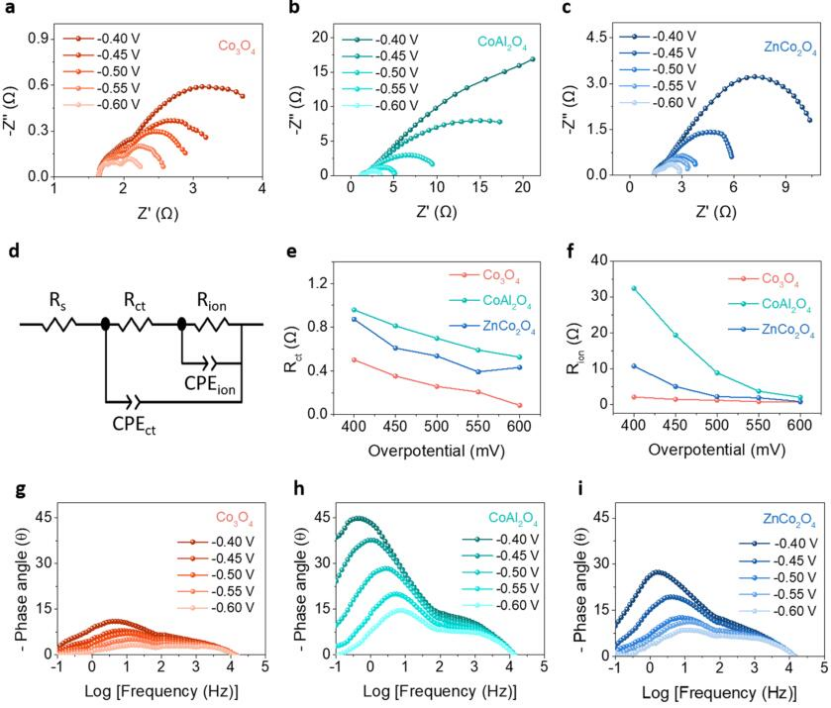
图3 a-c)催化剂在不同电位下的奈奎斯特图;d)模拟电化学响应的等效电路图;e,f)根据电化学阻抗模拟出的Rct和Rion;g-i)催化剂在不同电位下的波特相位图。
为了进一步探索碱性HER动力学,进行了阻抗谱(EIS)测试。图3a-c显示了催化剂的相应奈奎斯特图。为了精确量化这些反应系统的电荷和离子转移电阻,EIS结果采用图3d中的等效电路进行拟合。该模型由两部分组成:第一部分是Rs,代表电解质电阻,第二部分与电极-电解质界面反应有关,包括两组RC电路元件,其中一组包含CPEct和Rct,指的是双电层电容和法拉第反应过程中的电荷转移电阻。
另一组对应于电极-电解质界面上中间体的吸附行为,包括CPEion和Rion。 其代表中间吸附/解吸过程中产生的赝电容和离子转移电阻(图3e,f)。在不同的偏压下,Rct值遵循Co3O4
波特图(图3g-I)显示,在相同的偏压下,ZnCo2O4对应于比CoAl2O4更小的相位角,从而表明ZnCo2O4表面有更多的电荷参与法拉第反应,而不是储存在电极/电解质界面;而CoAl2O4则相反。这证明(Co3+-O)Oh是尖晶石型Co3O4 的HER活性的主要来源。
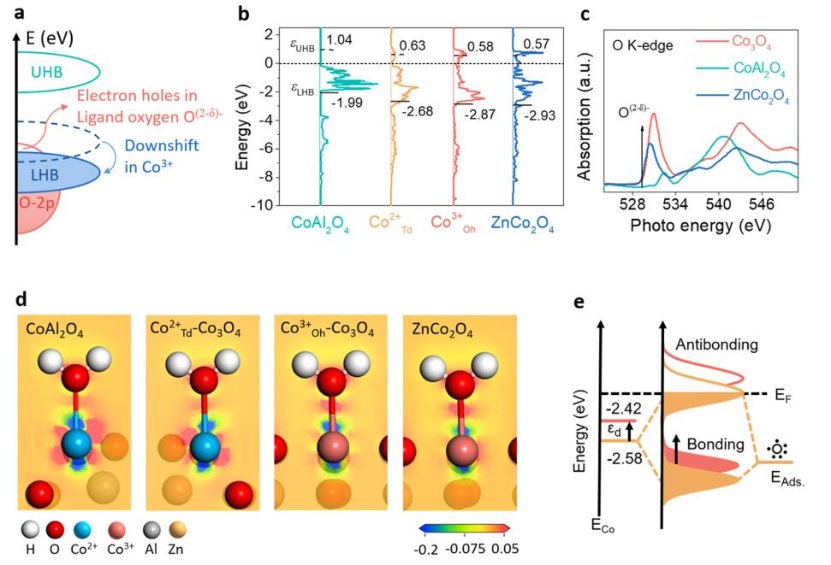
图4 a)Co3+诱发氧配体产生缺电子态;b)催化剂Co-3d轨道PDOS及其轨道中心;c)催化剂中O的K边XAS谱,用于表征氧配体的缺电子态;d)水分子在不同催化剂表面Co位点吸附的电子密度差分图;e)催化剂表面位点与反应物之间成键示意图。
为了阐明Cooh3+相对于CoTd2+的HER活性优势的来源,用密度泛函理论和XANES研究了d带结构。电子可能从氧配体的p带逃逸,从而产生定域空穴(记为O(2-δ)-)(图4a)。如图4b所示,低于和高于费米能级(EF)的带中心(εd)可以分别描述为LHB带中心(εLHB)和UHB带中心(εUHB)。可以定量地证实ZnCo2O4中CoOh3+的εLHB比CoAl2O4中的CoTd2+低得多,这有利于O(2-δ)-的形成。
为了进一步验证计算结果,通过O的K边XAS来检测表面氧配体的电子结构。如图4c所示,Co3O4和ZnCo2O4位于约529 eV处的肩峰可归因于氧配体中的局域空穴。CoOh3+诱导O(2-δ)-出现,削弱了其给电子能力,进一步削弱了氧配体对H*的吸附能力,最终促进了H2的脱附。水分子在Co3O4表面的CoOh3+和CoTd2+位置被吸收,然后计算出CoTd2+和CoOh3+相对于费米能级的d带中心分别为2.58和2.42 eV(图4e)。
由此得出结论,CoOh3+的反键态高于CoTd2+,这意味着表面CoOh3+与吸附水分子之间的相互作用增强,促进了二者之间的电子转移,从而提高了OH结合能,加速了水分子的解离。
总结与展望
本文作者通过在尖晶石Co3O4中进行选择性取代,在ZnCo2O4和CoAl2O4中分别构建了(Co3+-O)Oh和(Co2+-O)Td的孤立几何构型。证明了(Co3+-O)Oh是碱性HER活性的主要几何构型。CoOh3+的d带中心上移、相应氧配体的缺电子态出现分别有利于水的解离和氢气脱附,从而能够降低整个水还原反应的能垒。
这项工作阐明了尖晶石催化剂的几何构型对水还原活性的影响。通过构筑不同的几何构型,并分别研究其对催化活性的影响,能作为涉及多种活性位点的复杂催化反应的通用研究策略。
审核编辑:刘清
-
构建提高酸性水氧化催化剂稳定性的氧扩散路径2023-08-02 2028
-
催化剂分离和循环利用问题2023-05-23 1954
-
揭示卤素掺杂Sn基催化剂促进CO2电还原制甲酸盐原因2022-12-29 4239
-
如何提高HEAs催化剂的催化活性和优选设计研究2022-12-14 1935
-
“纳米岛”型催化剂突破传统催化剂活性和稳定性的矛盾2022-11-18 1807
-
应变效应对催化剂活性的影响2022-10-26 3667
-
析氢反应(HER)电催化剂在电解装置的广泛应用2022-09-28 12163
-
Mo配位FeCoNiMo碳负载高熵电催化析氧催化剂图文解析2022-09-20 3532
-
低结晶和异质结构AuPt-Ru@CNT像高效多功能电催化剂2022-05-31 781
-
高活性生物质碳负载Fe/Pt单原子双功能催化剂开发2021-02-12 3632
-
纳米笼催化剂保障燃料电池超长稳定运行时间2019-11-21 3657
-
碱性醇类燃料电池新型催化剂的研究2011-03-11 1987
-
锂离子蓄电池正极材料尖晶石型锰酸锂的制备2011-03-10 2777
-
脱氧催化剂纯化氢气的应用研究2009-01-10 1200
全部0条评论

快来发表一下你的评论吧 !
