

基于拓扑反应法制备高质量铁基超导薄膜
描述
一、薛其坤团队:基于拓扑反应法制备高质量铁基超导薄膜
背景介绍
制备高质量的铁基超导体并进行系统的物性表征是深入认识高温超导机理的重要途经。在FeCh(Ch=S, Se, Te)基超导体中,单层FeSexTe1-x/SrTiO3(0< x ≤ 1) 体系因其显著增强的超导电性和非平庸的拓扑性质而受到广泛关注。扫描隧道显微镜和角分辨光电子能谱等实验手段测得单层FeSexTe1-x的超导能隙闭合温度在50 ~ 83 K之间,远高于电输运测得的小于30 K的零电阻转变温度。
能隙闭合温度和零电阻转变温度之差异与薄膜的不均匀性存在密切联系。单层FeSe通常含有铁空位造成的密集的线缺陷,其超导能隙在线缺陷附近及线缺陷所包围的畴中受到显著抑制。单层FeSexTe1-x存在纳米尺度化学相分离,这可能会导致局域的磁有序。
这种样品的不均匀性既阻碍了高温超导机理的研究,又限制了单层FeSexTe1-x在量子计算相关研究方面的进一步探索。因此,优化单层FeSexTe1-x超导薄膜质量、提升其空间均匀性对基础研究和材料应用均具有重要意义。
相比于FeCh基超导体,FePn (Pn = P, As)基超导体具有更为庞大的家族体系(包括由Li、Ba/Sr、LaO和SmO等插层构成“111”,“122”和“1111”等体系)和更为丰富的物理特性(如磁有序结构等)和调控性。特别是,具有铁基块体材料最高超导转变温度55K的SmOFFeAs列于其中。
四方相结构的FePn为电荷不饱和态,实验上很难获得FePn超导表面。开展对FePn超导层的直接表征和调控研究具有与CuO2超导层研究相当的重要性和挑战性。获得四方相FePn层是开展相关表征和调控研究的关键性环节,对探索FePn基超导材料的配对机制以及提高FePn基材料的超导转变温度均具有重要意义。
成果简介
薛其坤团队通过拓扑反应法成功制备了系列的高质量单层FeSexTe1-x (0 < x ≤ 1)超导薄膜。研究团队首先利用分子束外延技术在SrTiO3(001)衬底上生长单畴的单层FeTe薄膜,然后将FeTe薄膜置于Se气氛下退火。Se的电负性强于Te,在适当条件下,Se可取代薄膜中的Te而形成单层FeSexTe1-x薄膜。精准调控衬底温度、Se束流及反应时间可实现Se化学配比x在0-1全范围精准控制。团队利用扫描隧道显微镜对单层FeSexTe1-x的电子态和表面形貌进行了原位表征,测得其超导能隙高达15 meV,并且Te和Se原子随机分布达到原子尺度化学均一。
特别地,与Fe-Se共沉积方法获得的单层FeSe相比,本方法制备的单层FeSe的线缺陷在数量和尺寸上均大幅减小,超导相干峰的强度显著增强。这些结果都表明拓扑反应法制备的FeSexTe1-x单层膜在纳米尺度上具有极佳的均匀性,为探究配对对称性、赝能隙等尚存争议的物理问题提供了研究载体。这项工作为合成和寻找亚稳相超导薄膜提供了研究范式。详见https://doi.org/10.1007/s12274-022-4718-3。
沿此途径,薛其坤团队通过多层FeTe薄膜与As原子的拓扑反应成功制备出了具有四方相结构的FeAs层,并在低温沉积钾原子后获得了KxFe2As2薄膜。团队利用扫描隧道显微镜对FeAs层和KxFe2As2薄膜的表面形貌和电子态进行了原位表征,发现FeAs层具有重构,而KxFe2As2膜具有沿重构对角线方向、周期为的条纹结构和~11 meV的超导能隙。
此外,输运测量结果显示这样制备的KxFe2As2/FeTe/STO样品的超导转变温度可达~ 10 K,相比体相KxFe2As2的3.8 K提高了一倍。由此,团队获得了界面增强超导特性的又一实例。更为重要的是,利用拓扑反应法成功制备具有四方结构的FeAs层为铁基超导薄膜的制备和调控提供了一个新途径,为探究铁基超导体的配对对称性等超导机制提供了研究载体。详见https://doi.org/10.1007/s12274-022-4956-4。
图文导读
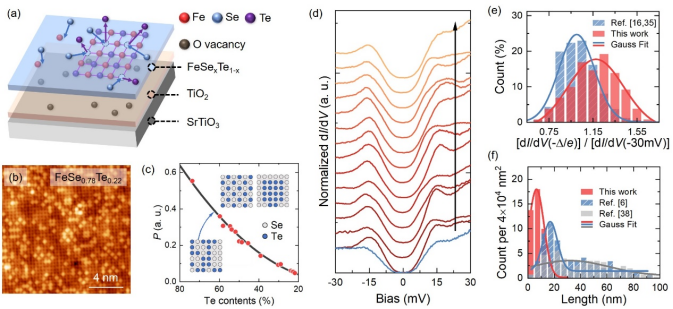
图1. (a)单层FeTe的硒化反应示意图。(b) 拓扑反应法制备的单层FeSe0.78Te0.22的原子分辨图。(c) 不同Te含量样品的参量P的值(红色圆点),其中P为最近邻Te-Te化学键数目与阴离子间总化学键数目之比,黑色曲线为Te原子随机分布时P与Te含量的依赖关系。(d) 拓扑反应法制备的单层FeSe的隧道谱,其相干峰高度相较于共蒸发法情形(蓝色)有显著提升。(e) 相干峰高度统计及与共沉积结果对比。(f) 线缺陷数量和长度统计及与共沉积结果对比。
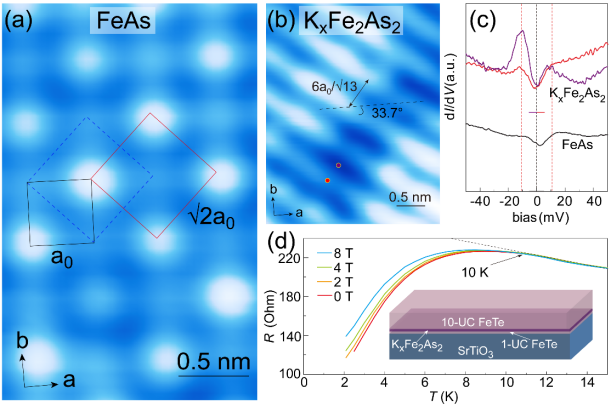
图2. (a) 拓扑反应法制备的FeAs层的原子分辨图显示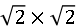 重构。(b) KxFe2As2表面形貌图显示沿
重构。(b) KxFe2As2表面形貌图显示沿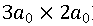 重构对角线方向、周期为
重构对角线方向、周期为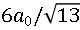 的条纹相。(c) FeAs和KxFe2As2表面的扫描隧道谱线。KxFe2As2表面的扫描隧道谱线显示± 11 meV处相干峰强度随条纹结构调制变化。(d) KxFe2As2/FeTe/STO样品的结构示意图和R-T测试结果。
的条纹相。(c) FeAs和KxFe2As2表面的扫描隧道谱线。KxFe2As2表面的扫描隧道谱线显示± 11 meV处相干峰强度随条纹结构调制变化。(d) KxFe2As2/FeTe/STO样品的结构示意图和R-T测试结果。
二、北航贾彬彬/北理工陈卓老师:促进电化学CO2还原的非对称Ni-P1N3活性中心的合理设计
背景介绍
利用可持续能源发电将二氧化碳(CO2)催化转化为增值产品正受到越来越多的关注。单位点催化剂(SSC)由于其高活性中心和高原子利用率,在电催化CO2还原反应(CO2RR)中具有优异的性能。从实验和理论层面来看,贵金属SSC(如Au、Ag和Pd)对CO2RR寄予厚望。但其稀有性和高昂的价格限制了其实际应用。同时,据报道,Fe、Co、Ni、Mn和其他非贵过渡金属(TM)SSC在CO2RR中也显示出高催化活性。
对于蓬勃发展的碳基TM SSC,提出金属氮(M-Nx)位点可能是有利于CO2吸附和还原反应的活性中心。与中心原子配位的掺杂N的数量和类型的调整将导致过渡金属电子排列的差异,这将影响CO2RR中间产物的吸附和解吸。否则,还可以通过用外来的元素替换部分N原子来改变配位微环境,例如磷(2.19),与N(3.04)和C(2.55)相比,磷具有更大的半径和更低的电负性。P的加入改变了原始M-N键的长度和中心金属原子的正电荷。
研究方法
通过热解的MOF框架构建了界面调控的P、N双配位的碳基负载的镍单位点催化剂(表示为Ni-PxNy,x=1,2和y=3,2),应用于电催化CO2还原反应(CO2RR)中。
成果简介
引入同步辐射XAS测试确定Ni中心原子的局域配位结构。实验和理论结果表明,Ni-PxNy单分散活性位点是高催化活性的根源。其中Ni-P1N3上的CO电流密度显著高于未经P改性的Ni-N4催化剂,在-0.65至-0.95 V(vs RHE)的宽电位范围内具最高FECO,为85.0-98.0%,同时反应20小时后仍能保持初始FECO的94%。实验和理论结果表明,Ni-P1N3催化剂具有与COOH*和CO*关键反应中间体适度的结合强度,因而具有更高的催化CO2还原活性。
图文导读
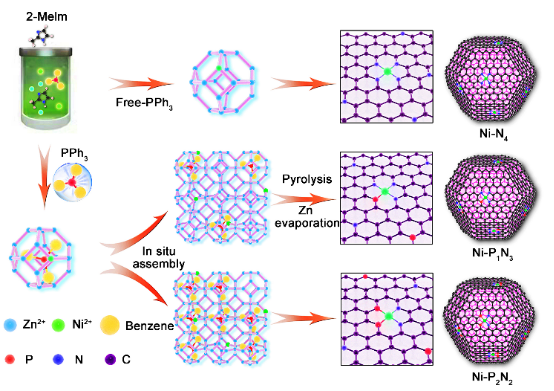
图1. Ni-PxNy样品的制备流程示意图
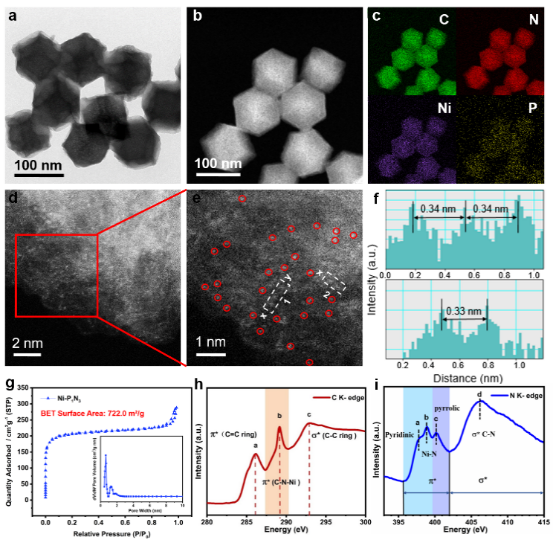
图2. Ni-P1N3的(a)TEM图像;(b)STEM图像和(c)相应的EDS图像;(d)和(e)HAADF-STEM及其放大图像;(f)沿e中X-Y线的强度分布;(g)BET表面积和孔径分布;(h)Ni-P1N3的 C K-边XANES光谱;(i)N K-边XANES光谱 图2c中的图像上彩色亮点证实了样品中Ni、P、N和C物种的存在,表明Ni、P和N元素成功掺入且均匀分布在整个碳基结构中。
图2d、2e上独立分散的白色亮点为镍金属原子,整个碳基材料上并未观察到镍金属纳米颗粒的存在,表明Ni金属原子呈高度单分散状态。测量图2e上镍金属白色亮点间的距离,沿X-Y的相应强度分布表明(图2f),单个Ni原子之间的间距至少为0.33 nm,明显大于单个Ni原子的有效半径(124.6 pm = 0.125 nm),进一步验证了Ni物质在制备的Ni-P1N3中的孤立特性。
图3. Ni-PxNy及标准样品的Ni K边(a)XANES和(b)EXAFS光谱;Ni-P1N3的(c)k空间和(d)FT-EXAFS拟合曲线;(e)Ni-P2N2和(f)Ni-N4的FT-EXAFS拟合曲线;(g)Ni箔、Ni-P2N2、Ni-P1N3和Ni-N4的WT-EXAF图 基于同步辐射的XAS测量深入探究原子级Ni SSCs的界面结构。
Ni物种的平均氧化态可以通过Ni K边的吸收阈值描述。在XANES曲线中(图3a),Ni-P1N3的位置在Ni-N4和Ni-P2N2之间,表明Ni的氧化态在二者之间。通过对比Ni SSCs样品与标准样品的傅里叶变换(FT)EXAFS光谱,探究Ni-P1N3、Ni-N4和Ni-P2N2的界面结构,如图3b所示。Ni-P1N3样品在1.44 Å处有一个明显的FT峰,对比仅含有Ni-N配位的NiPc可知,该峰主要来自Ni-N配位的散射;在1.89 Å处检测存在一个肩峰,与NiP2的FT-EXAFS光谱进行比较,表明该信号来自Ni-P散射,证实了Ni-P键的形成。
此外,与Ni箔相比,在Ni-P1N3、Ni-N4和Ni-P2N2谱图上均未观察到源自Ni-Ni的散射峰,证实催化剂上不含有Ni金属小颗粒,Ni主要以单个原子的形式存在于碳基材料上,同时与N和P原子形成配位结构。进行小波变换处理(图3 g)进一步验证Ni的原子色散,与Ni箔相比,Ni-P1N3、Ni-N4和Ni-P2N2谱图均未在对应位置出现波峰。对Ni-P1N3进行EXAFS拟合定量提取Ni K边结构参数,拟合结果如图3c-f所示。
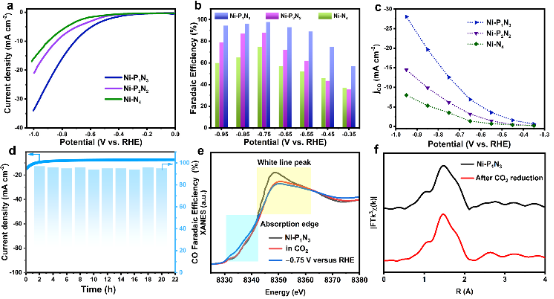
图4. Ni-PxNy及标准样品的催化性能和原位XAFS表征。 我们使用三电极H电池评估了CO2饱和0.5 M KHCO3溶液中的CO2RR性能。在线性扫描伏安法(LSV)曲线中(图4a),Ni-P1N3在-1.0 V时的电流密度为33.98 mA cm-2,而RHE的电流密度远大于比较的Ni-P2N2(21.82 mA cm-2)和Ni-N4(16.97 mA cm-2)。
此外,当在Ar饱和电解质中测量Ni-P1N3时,图S19中检测到电流密度显著降低,起始电位增加,表明活性来源于CO2RR。通过气相色谱法和核磁共振波谱法对产物进行了测量,表明在CO2RR过程中仅生成CO和H2。图4b显示了Ni SSC的CO法拉第效率(FECO)。其中,Ni-P1N3在整个电位区间内超过Ni-P2N2和Ni-N4,在-0.75 V时达到98%的最大FECO。此外,图4c中显示了CO电流密度(jCO)与外加电位的关系。在-0.75 V的电位下,Ni-P1N3显示出14.30 cm–2的高电流密度,是Ni-P2N2和Ni-N4的2.1倍和3.6倍。
此外,Ni-P1N3也表现出良好的稳定性,在电解20 h后,相对于初始FECO,其保持94%(图4d)。 为了深入研究CO2RR过程中催化剂的演化,对反应前后的催化剂进行了原位X射线吸收光谱测试。近边光谱(图4e)显示,反应条件下Ni-P1N3的吸收边缘向较低能量转移,表明Ni的价态在反应过程中略有下降。对反应前后的结构数据进行对比(图4f),反应后催化剂的结构没有发生明显变化,仍保留着Ni-P1N3四配位的界面结构,具有一定的结构稳定性。
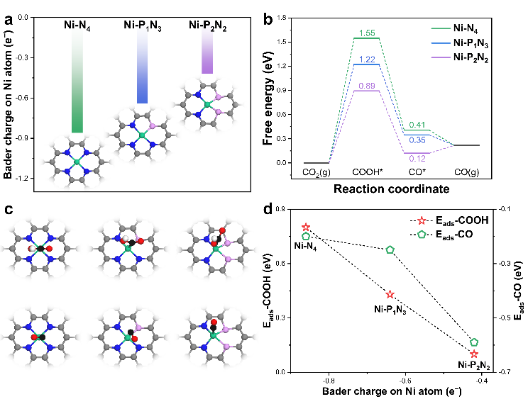
图5. (a)Ni-N4、Ni-P1N3和Ni-P2N2催化剂的原子结构以及中心Ni原子上的Bader电荷计算值;(b)CO2RR自由能图;(c)反应中间体的原子结构示意图(d)COOH*和CO*在中心Ni原子的吸附能(Eads) 为分析Ni-N4、Ni-P1N3和Ni-P2N2催化剂间性能差异的来源,首先计算不同原子界面结构中Ni位点到碳基底的Bader电荷转移数,如图5a所示,随着Ni-N4结构中N原子逐渐被P原子取代,从Ni活性中心转移到碳基底的电子数量逐渐减少。因此,具有P配位的Ni活性位点保留了更多的电子,这可能会大大增强与吸附物的相互作用。
计算分析Ni-PxNy上CO2RR生成CO的自由能差异,如图5b。正如之前研究所示,在CO2电催化生成CO通常由两个步骤决定,即COOH*中间体的形成和CO*的解吸。因此,理想的CO2RR-to-CO催化剂应与COOH*形成强结合,但同时与CO*结合较弱。然而,目前的工作中COOH*和CO*之间存在线性比例关系,为了设计高效的催化剂,规避反应中间体之间比例关系对于设计高效催化剂是必要的。
图5b的例子中,在Ni-N4催化剂中添加P原子后,COOH*形成自由能从1.55 eV(Ni-N4)逐渐下降到1.22 eV(Ni-P1N3),然后下降到0.89 eV(Ni-P2N2),表明COOH*中间体的结合强度随着P原子的掺入逐渐增强。但伴随着对COOH*中间体吸附结合的增强,Ni活性位点上对CO*结合强度也随之提升,不利于CO的解吸,并最终限制了CO2RR的催化性能。
反应中间体增强的结合强度与Ni活性位点转移到基底的电荷量有关(图5d)。反应中间体(即COOH*和CO*)的原子结构及其吸附示意图(如图5c)。Ni-N4、Ni-P1N3和Ni-P2N2催化剂上COOH*结合能以相同的幅度增加(0.33 eV),而Ni-P2N2催化剂对CO的吸附强度显著增加,并从Ni-N4和Ni-P1N3的物理吸附转变为化学吸附,表明CO在Ni-P2N2催化剂上的解吸过程缓慢。
因此,在Ni-N4上,CO2RR的性能受到COOH*产生的限制,而Ni-P2N2的性能则更多受CO*脱附的影响。显然,在这种情况下,CO分子稳定地吸附在Ni原子上,不利于CO2RR过程的进行。在这方面,具有中等COOH*和CO*中间体结合强度的Ni-P1N3催化剂则是CO2RR转化为CO的最理想催化剂,COOH*的形成和CO*解吸都不会过于限制整个反应过程的限制。DFT结果与实验测试结果一致,Ni-P1N3表现出比Ni-N4和Ni-P2N2增强的CO2RR活性。
三、张袁斌课题组:具有柔性筛分效应的新型阴离子柱撑MOF用于从CO2和CnH4中高效捕获C2H2
背景介绍
乙炔(C2H2)是工业上生产各种必需化学品和聚合物的主要原料。它是由天然气的部分燃烧或碳氢化合物的裂解产生的,其中二氧化碳(CO2)和其他C1 -C2碳氢化合物会与其共存,需要去除以生产高纯度的C2H2。目前,采用能耗较高的低温蒸馏和溶剂萃取法从其他气体中提取C2H2。由于沸点相近,这些方法的效率低、能耗高且对环境有污染。
因此,基于多孔固体吸附剂的物理分离方法由于较低的成本和能耗而引起了人们的关注。然而,这些气体分子在分子大小(动力学直径:C2H2和CO2均为3.3Å,CH4为3.8Å,C2H4为4.2Å)和物理性质方面的相似性也加大了分离的难度。金属有机框架(MOFs)以其强大的结构可预测性和对孔隙大小/形状和功能的可调性而被广泛关注。
研究方法
慢扩散方法
ZNU-4的合成方法:在一个5mL的玻璃管中,在最下层加入1mL含有(NH4)2TiF6(1mg)和Cu(NO3)2‧3H2O(1mg)的水溶液,中间加入2mL MeCN/H2O(1:1)的缓冲溶液,最上层加入1 mL DIB的MeCN溶液。然后将玻璃管静置放置在室温下,过几天会在中间层看到有蓝色块状晶体产生。
ZNU-5的合成方法:在一个5mL的玻璃管中,在最下层加入1mL含有(NH4)2TiF6(1mg)和Cu(NO3)2·3H2O(1mg)的水溶液,中间加入3mL MeOH/H2O(1:1)的缓冲溶液,最上层加入1 mL DIB的MeOH溶液。然后将玻璃管静置放置在室温下,过几天会在中间层看到有紫色针状晶体产生。
成果简介
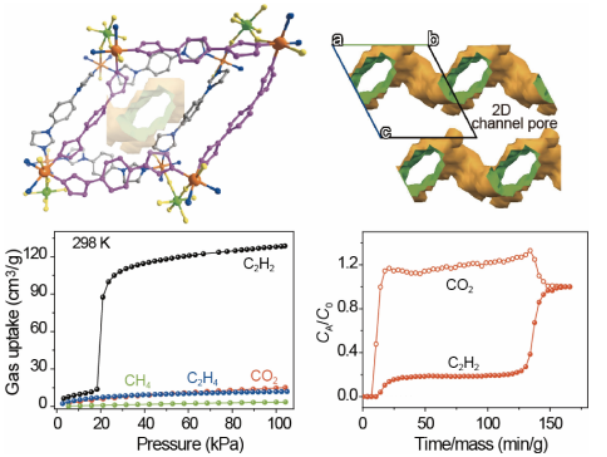
基于以上思考,我们报道了一种新的TiF62−阴离子(TIFSIX)柱撑的金属有机框架材料ZNU-5 (ZNU =浙江师范大学),该材料可通过门开产生的分子筛分效应,以低吸附热选择性捕获C2H2。ZNU-5在1.0 bar和298 K时吸附大量的C2H2(128.6 cm3 /g), 但对CO2、CH4和C2H4的吸附微乎其微。如此高的吸附量在分子筛分的MOFs中还没有实现过。
穿透实验进一步证实了该材料可从多组分混合气体中高选择性地捕获C2H2。ZNU-5可分别从等摩尔C2H2/CO2、C2H2/CO2/CH4和C2H2/CO2/CH4/C2H4混合物中捕获3.3、2.8和2.2 mmol/g的C2H2。此外,2.6、2.0和1.5 mmol/g,且纯度> 98%的C2H2可以从解吸过程中回收。ZNU-5结合高的吸附容量、低吸附热、良好的可回收性,具有良好的C2H2纯化前景。
图文导读
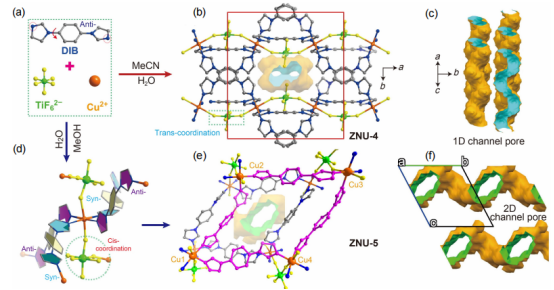
图 1 (a) ZNU-4和ZNU-5的结构组成单元。(b) ZNU-4的多孔结构。(c) ZNU-4的孔隙。(d) ZNU-5中的配位模式。(e) ZNU-5的多孔结构。(f) ZNU-5的孔隙。孔隙由半径为1.2 Å 的探针进行测试。
要点:图1a是ZNU-4和ZNU-5的组成单元,两种材料合成方法的差异在于慢扩散过程中使用溶剂的不同。ZNU-4是由MeCN/H2O溶剂参与慢扩散形成的,而ZNU-5是由MeOH/H2O参与的慢扩散所获得的。图b是ZNU-4的框架结构,在ZNU-4中每个Cu(II)离子与来自四个不同DIB配体的四个咪唑氮原子和来自两个TiF62-基团的两个氟原子相连。
其中,DIB配体为anti-构型,而TiF62-通过trans构型配位。6个Cu2+和6个DIB配体产生一个非常扭曲的环。每个单元中都包含两个相邻的狭窄的一维通道,表面装饰着氟原子。图d是ZNU-5的框架结构,其中Cu(II)为八面体配位,连接四个DIB配体(一半采取syn-构型,一半采取anti-构型)和两个TIFSIX阴离子(采取cis-构型),形成具有pcu拓扑结构的非互穿框架。与ZNU-4不同,ZNU-5具有二维的孔隙通道。
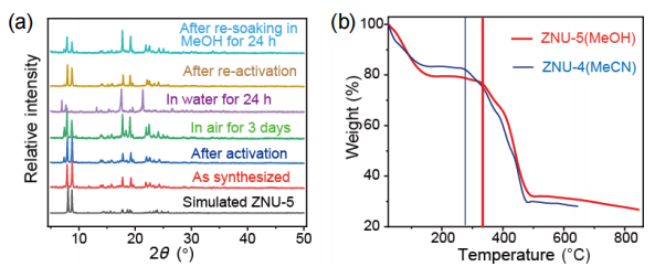
图2(a)不同条件下ZNU-5的PXRD图。(b)ZNU-4和ZNU-5的热重分析曲线。 要点:从PXRD的测试结果可以看出制备的ZNU-5具有很好的相纯度,且在活化后和空气中放置三天,材料依旧稳定。但将该材料浸泡在水中24h后,得到了新的相,但将水中浸泡的样品再次活化或浸泡甲醇后,依旧可以回到合成的相状态,表明其相的转变具有可逆性。从热重的测试结果可以看出,ZNU-4的热稳定性在290℃,ZNU-5较ZNU-4的热稳定性高,为340℃。
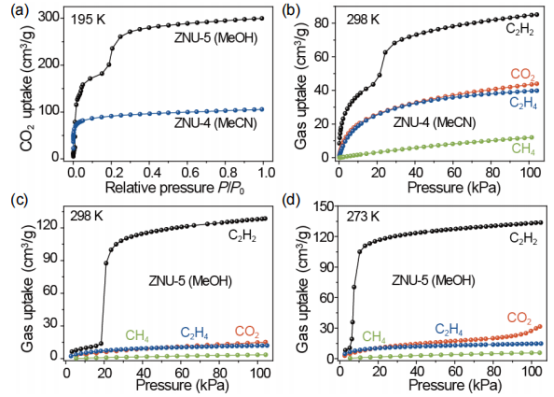
图3 (a) ZNU-4和ZNU-5在195 K时的CO2吸附等温线。(b)在298 K时,ZNU-4中C2H2、CO2、C2H4和CH4的吸附等温线。(c,d)在298K和273 K时,ZNU-5中C2H2、CO2、C2H4和CH4的吸附等温线。 要点:图a为195K下ZNU-4和ZNU-5的吸附曲线。测试结果显示ZNU-4的吸附呈现I型吸附等温线,在100 kPa时最大吸附量为105.7 cm3 g-1,BET表面积为358.6 m2·g-1。
与ZNU-4不同,ZNU-5的CO2吸附过程可分为两个阶段,第一步和第二步的CO2吸附量分别为200.9 cm3g-1(15 kPa)和299.8 cm3g-1(100 kPa),BET表面积为751.5 m2·g-1。计算得到的孔径为5.2 Å。气体吸附等温线显示,ZNU-5在298K时对C2H2存在门开效应,而对C2H4、CH4和CO2却没有该现象。
其在298 K和100 kPa时表现出较高的C2H2吸附量为128.6 cm3g-1,比ZNU-4(85.1 cm3·g-1)的吸附量高51.1%。与此同时,在298 K时,当压力高达100 kPa时,ZNU-5完全阻止了CO2、C2H4和CH4的进入。因此,在100 kPa下较高的C2H2吸附能力,以及较低的CO2、C2H4和CH4吸附,使ZNU-5具有从C2H2/CO2/C2H4/CH4四组分混合物中实现一步纯化C2H2的能力。
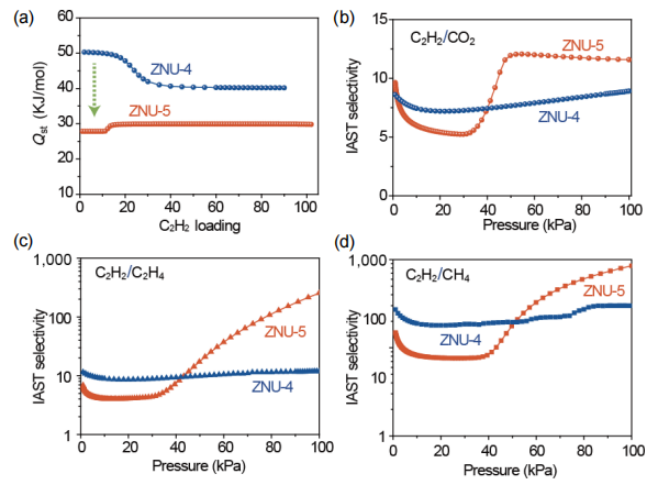
图4 (a) 在ZNU-4和ZNU-5中C2H2的Qst。(b-d)在298K下,ZNU-4和ZNU-5对等摩尔C2H2/CO2、C2H2/C2H4和C2H2/CH4混合物的IAST选择性。
要点:图a为ZNU-4和ZNU-5对C2H2的Qst值。其中,ZNU-5的C2H2Qst值(27.8 kJ·mol-1)远低于ZNU-4(50.3 kJ·mol-1),说明其再生能耗相对较低。图b、c和d为ZNU-4和ZNU-5对C2H2/CO2、C2H2/C2H4和C2H2/CH4的IAST选择性比较。由于ZNU-5中存在对于C2H2的门开效应,使其IAST选择性较ZNU-4有所提升。ZNU-5在100 kPa时具有较高的选择性,分别高达12、255和850,优于ZNU-4的选择性,分别为9、12和300。
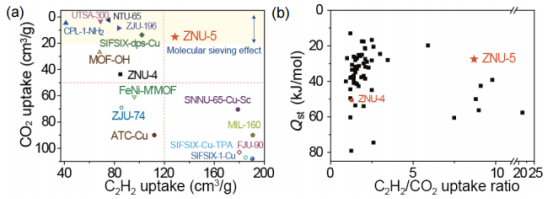
图5(a) ZNU-4/ZNU-5和其他材料在100 kPa和298K时,C2H2和CO2吸附量的比较。(b) ZNU-4/ZNU-5与其他材料对C2H2的Qst及C2H2/CO2的吸附容量比比较。 要点:通过与其他材料对比,ZNU-5显示出非常高的C2H2吸附和非常低的CO2吸附,这也反映在图5b中的C2H2/CO2吸附比中。此外,ZNU-5是极少数具有C2H2/CO2吸收比>8且Qst值< 30 kJ/mol的多孔材料。
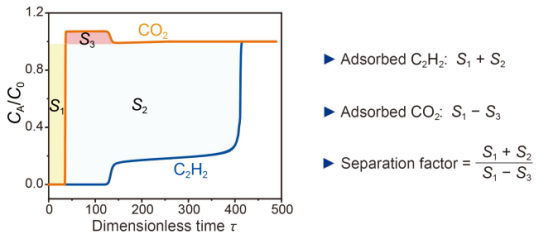
图6ZNU-5在298 K下对C2H2/CO2(50/50)的突破曲线。
要点:瞬时穿透测试评估了ZNU-5对等摩尔C2H2/CO2(50/50)混合物的分离性能。如图6所示,ZNU-5对C2H2呈阶梯式穿透曲线,这在刚性吸附剂中从未观察到过。通过对曲线的分析可看出捕获的C2H2量仍然很大,而CO2量可以忽略不计,从而得到了一个较高的分离系数。此外,C2H2需要解吸过程才能获得,因此穿透过程中的轻微泄漏不会对C2H2的动态捕获有很大的影响。
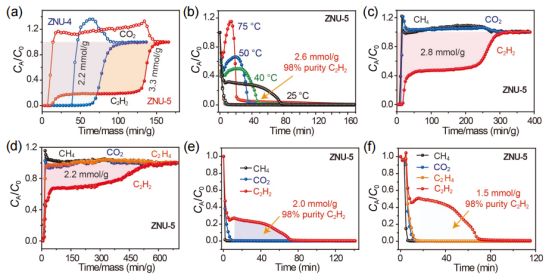
图7 (a) ZNU-4和ZNU-5的C2H2/CO2 (50/50)穿透曲线。(b)ZNU-5在25℃、40℃、50℃和75℃时的C2H2/CO2 (50/50)解吸曲线。(c)在ZNU-5中,C2H2/CO2/CH4 (33.3/33.3/33.3)穿透曲线。(d)在ZNU-5中, C2H2/CO2/C2H4/CH4(25/25/25/25)分离的穿透曲线。(e)ZNU-5中C2H2 /CO2 /CH4 (33.3/33.3/33.3)分离的解吸曲线。(f)ZNU-5对于C2H2/CO2/C2H4/CH4(25/25/25/25)的解吸曲线。
要点:为了评估实际的分离性能,并确定阶梯式穿透现象,进行了实验穿透测试。实验结果表明,实验穿透曲线与模拟结果非常相似。对于等摩尔的C2H2/CO2混合物,ZNU-5可以实现有效的分离,可以获得3.3 mol·g-1的C2H2,而ZNU-4吸附C2H2的量仅为2.2 mol·g-1。随后,研究了不同的解吸温度对再生的影响。从图7b可以看出,随着解吸温度的升高,C2H2的解吸时间逐渐缩短。
在解吸温度为25℃时,吹出CO2后,可从柱中回收2.6 mol·g-1纯度为98%的C2H2。后续的三组分及四组分穿透测试仍然得到了高纯度的C2H2,证明了ZNU-5仍具有较好的从三组分和四组分混合物中得到高纯C2H2的能力。
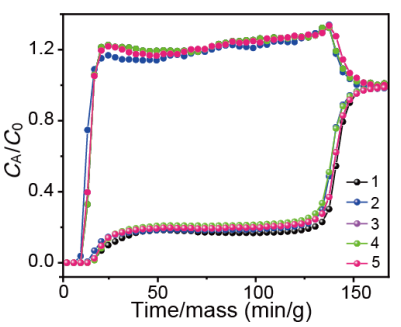
图8 C2H2/CO2(50/50、v/v)混合物的动态穿透曲线的五次循环。
要点:在五次循环穿透测试中,ZNU-5材料的穿透时间及容量均没有发生变化。证明该材料是一种很有前途的吸附剂,具有良好的循环稳定性。
四、广东工业大学易国斌、林霄峰课题组:弹性模量可控的自擦除动态表面图案助力多重信息存储与抗篡改应用
背景介绍
近年来,动态表面图案(DSPs)由于其对编码表面功能和性质的按需控制而得到了迅速发展,有望应用于光电器件、智能窗户、防伪,尤其是在信息安全方面。通过将智能材料引入褶皱以响应外部刺激(例如湿度、光、温度、pH 和拉伸应力),可以制造可控和响应迅速的 DSP,并针对表面形貌和功能进行动态调整。
相比单模式、双模式,甚至多模式的光学图案化技术,动态表面图案可以有效提高信息存储容量和加密能力。尽管许多研究报告了基于褶皱和荧光的双模式 DSPs发展,用以加密复杂信息,但由于 DSP 中不可控的褶皱保留时间和相互干扰的皱纹和荧光状态,应用于供应链的防篡改仍然是一个非同寻常的挑战。
因此,本研究旨在开发和设计一种具有快速响应的新型DSPs,可以控制褶皱保留时间并独立调节褶皱和荧光状态中的不同信息。这种新策略可以为产品安全或无墨印刷中的防篡改、高密度和多编码信息存储提供一种新方法。
成果简介
本研究开发了一种基于可控弹性模量的图案化策略,以制造具有更高信息存储密度的自擦除动态表面图案 (S-DSPs)。这些新型 S-DSPs 战略性地将氨基共聚物(ACO、DMAEMA-co-DMAPAA)与 9-蒽甲醇 (9-AM) 作为表层,聚二甲基硅氧烷 (PDMS) 作为柔性基板,设计成双层多编码体系。它可以携带几种不同类型的信息,如褶皱和荧光。
为了赋予皮肤层更宽范围的弹性模量,我们通过甲基丙烯酸 2-(二甲氨基)乙酯 (DMAEMA) 和 N, N-二甲氨基丙基丙烯酰胺 (DMAPAA) 的自由基聚合合成了相对低分子量的共寡聚物(ACOs),同时皮肤层中9-AM的羟基和ACOs的酰胺基会进行结合形成氢键构成超分子交联网络。
随后,双层系统中压缩应变和临界褶皱应变之间的差异可以通过皮肤层的光二聚作用来精确调节,从而使 S-DSPs 产生快速响应(最小< 1 分钟)和可自擦除的褶皱图案(3 分钟~8 天)。此外,荧光图案可以在不改变褶皱图案的情况下独立地被擦除和重新编程,从而开发出具有多种编码信息存储能力的 S-DSPs。
同时,具有可控自擦除功能的S-DSP还可用于产品的保质期,极大地提高了供应链的防篡改能力。据我们所知,这是控制褶皱保留时间并独立调节褶皱和荧光状态中不同信息的DSPs的第一份报告,该策略可以应用于防篡改、高密度和高编码信息存储的产品安全、智能显示器和无墨打印。
图文导读
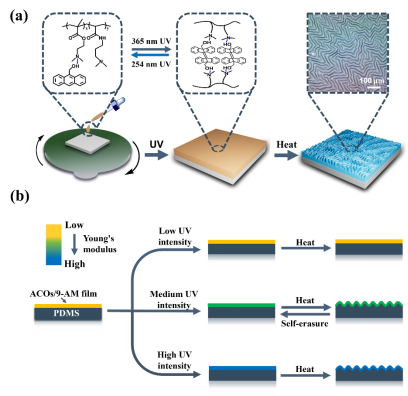
图1 S-DSPs的调控策略。(a) 制备 DSPs的策略。(b) 三种不同紫外线强度下起皱变化的示意图。
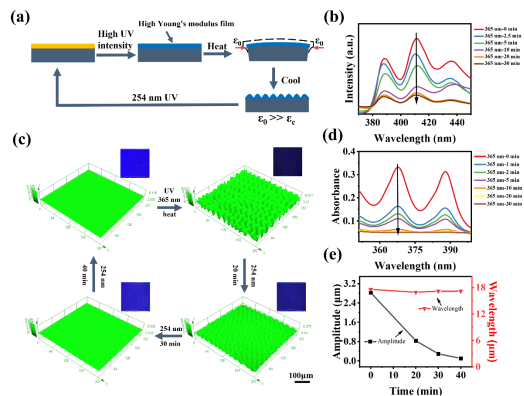
图 2 褶皱和荧光同时变化的 DSPs。(a) 高强度紫外线照射的起皱动力学。(b) ACOs/9-AM 薄膜在 365 nm 紫外光下不同照射时间的荧光光谱。(c) 样品在 365 nm 紫外线照射后通过热处理起皱和在 254 nm 紫外线照射下消除皱纹的 LSCM 图像。插图是相应的荧光图像,比例尺:100 μm。(d) ACOs/9-AM 薄膜在 365 nm 紫外光下不同照射时间的紫外-可见光谱。(e)褶皱擦除过程中波长和幅度的详细变化。

图 3 在紫外线照射下通过光掩模形成的褶皱和荧光图案。(a) 在自然光和紫外光下未处理样品的图像。放大图像显示样品表面透明且无褶皱,比例尺:5 mm。(b) 365 nm 紫外线照射下的“蝴蝶”褶皱和相应的荧光图案照片(紫外线强度:1000 mW·cm-2)。放大的图像显示了样品内部褶皱的无序形状,比例尺:100 μm。(c) 带有“条纹”光掩模,在 254 nm 紫外线照射下褶皱消除和荧光增强的图案。放大的图像显示了擦除的细节,比例尺:5 mm。

图 4 仅有荧光变化的 DSP。(a) 低强度紫外线照射的起皱动力学(紫外线强度:250 mW·cm-2)。(b) 低强度紫外线照射后再高温加热处理的图案变化。比例尺 1:5 mm 比例尺 2:4 mm。(c) Ⅰ在高紫外线强度(紫外线强度:1000 mW·cm-2)的照射下形成褶皱和荧光的“工”图案;Ⅱ高温加热后“工”的荧光图案消失;Ⅲ低强度紫外线照射后的荧光图案发生变化。比例尺 1: 5 mm比例尺 2:4 mm。
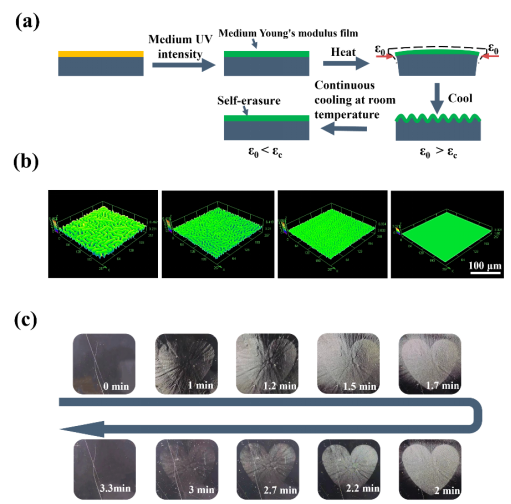
图 5 具有可自擦除褶皱的 DSPs。(a) 中等强度紫外线照射的起皱动力学(紫外线强度:600 mW·cm−2)。(b) 中等强度 365 nm 紫外线照射下褶皱自擦除的 LSCM 图案,比例尺:100 μm。(c)随着时间的推移褶皱自我擦除的光学图像。
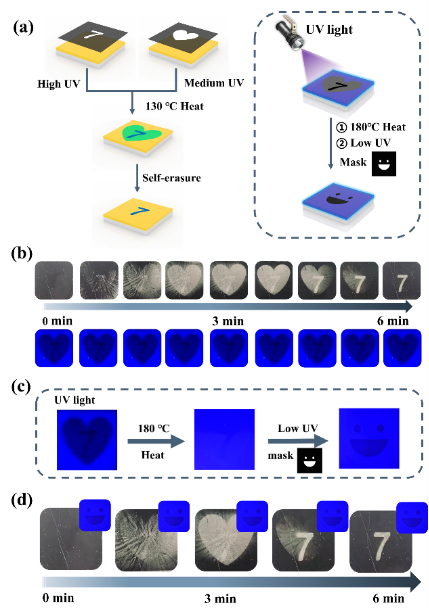
图 6 用于多编码信息存储的 S-DSPs。(a) 多编码 S-DSPs的设计思路。(b)在自然光下具有褶皱变化的 S-DSPs的光学图像。(c) 荧光图案重构的过程。(d) S-DSPs 的褶皱和荧光变化。
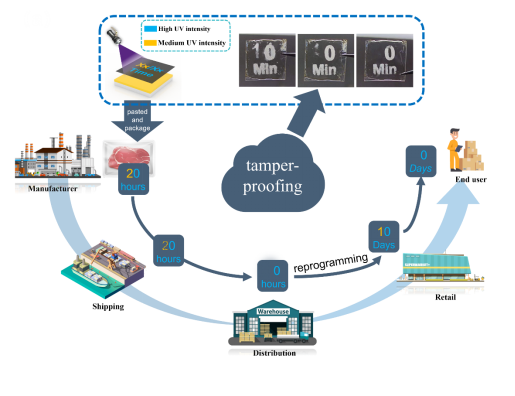
图 7 S-DSPs 的可控自擦除在供应链中防篡改应用。
审核编辑:刘清
-
protel输出高质量gerber2011-12-12 26423
-
高质量C++、C编程指南2012-08-06 5129
-
高质量C&C++2012-08-16 2754
-
高质量C语言编程2013-07-22 14054
-
编写高质量C语言代码2013-07-31 4781
-
林锐《高质量C语言编程》2013-08-17 2473
-
高质量编程2016-02-27 7301
-
【下载】高质量干货-22本高质量EMC电磁兼容性设计资料2020-03-20 6003
-
请问怎么才能设计出高质量的印制线路板?2021-04-23 1536
-
高质量的双量子比特门操作2021-07-29 1515
-
PVDF/PDDA静电自组装超薄膜的制备与表征2009-02-27 1580
-
铁基颜料铁黄制备工艺流程2009-03-30 2244
-
中科院研究所宣布:世界首根百米级铁基超导长线研制成功2018-04-24 819
-
铁基超导体.重费米子化合物.赝能隙.这些量子材料的概述2018-04-02 11203
-
铁基超导基本层状结构单元2022-10-21 2978
全部0条评论

快来发表一下你的评论吧 !
