

平衡配体效应和应变效应提升Pt合金的ORR活性
描述
01
导读
氢燃料电池和金属空气电池等电化学装置是实现具有环境友好的能源供应和储存道路的关键技术之一。近来,氧还原反应(ORR)作为这几种能量供应和存储装置的重要组成部分,被得到重点研究。鉴于人们普遍认为配体和应变效应对铂(Pt)合金优异的ORR性能起着关键作用,但其活性位点的性质仍不清楚。为此,需要深入了解带电固/液界面的电化学过程,其中原位技术和操作技术结合计算模型被认为是最适合评估活跃区性质的方法。
02
成果简介
近日,权威期刊Energy & Environmental Science上发表了一篇题为“A trade-off between ligand and strain effects optimizes the oxygen reduction activity of Pt alloys”的文章。该工作基于电化学扫描隧道显微镜(EC-STM)的原位技术,在纳米水平分辨率的酸性介质中“可视化”了Pt3Ni(111)上的活性位点。
结果表明,与纯Pt相比,Pt3Ni(111)面的活性中心位于台阶位附近的凹区,而平台位的活性与纯Pt相当甚至更低。此外,作者还通过一个基于合金和应变敏感广义配位数的模型证实了实验结果。本文结合理论和实验工具为设计更高效的Pt合金氧还原电催化剂提供了策略。
03
关键创新
(1)本文结合实验原位扫描探针技术和涉及形态和成分敏感描述符的计算模型阐明铂合金催化剂上的活性位点,并说明了Pt与三维过渡金属(Ti, Co, Ni, Cu)合金化所产生的配体效应和晶格压缩相互作用逐渐增加了表面Pt原子的广义配位数,从而使(111)阶高度活跃;
(2)本文基于合金和应变敏感广义配位数的模型能够评估各种Pt合金催化剂上最佳催化活性位点的组成和几何构型。
04
核心内容解读
本文中,作者使用实验技术对Pt3Ni(111)表面进行研究,而为了保证模型体系的定义明确,采用表面科学和电化学技术对Pt3Ni(111)单晶表面进行了表征。图1a的典型LEED模式表明,从衍射光斑的六边形排列,确认了表面的(111)方向,其中没有证据表明结构改变了表面的周期性。此外,在对新制备的晶体表面进行典型LT-STM(图1b)测量后,其(111)表面可以通过原子分辨率区分。
在0.1 M HClO4中测试了Pt3Ni(111)表面的电化学性能。图1c所示的伏安曲线与文献一致,其存在典型的表面氧化和还原峰。在小于0.3 VRHE时,特征可归为氢的吸脱附,而在0.8 VRHE附近的特征可归为OH的吸脱附。在旋转圆盘电极(RDE)实验中评估了ORR性能(图1d),催化剂在0.9 VRHE下获得了7.6 mA cm-2(归一化几何面积)的比ORR活度。这比以前的研究中报告的室温下的活性要高。
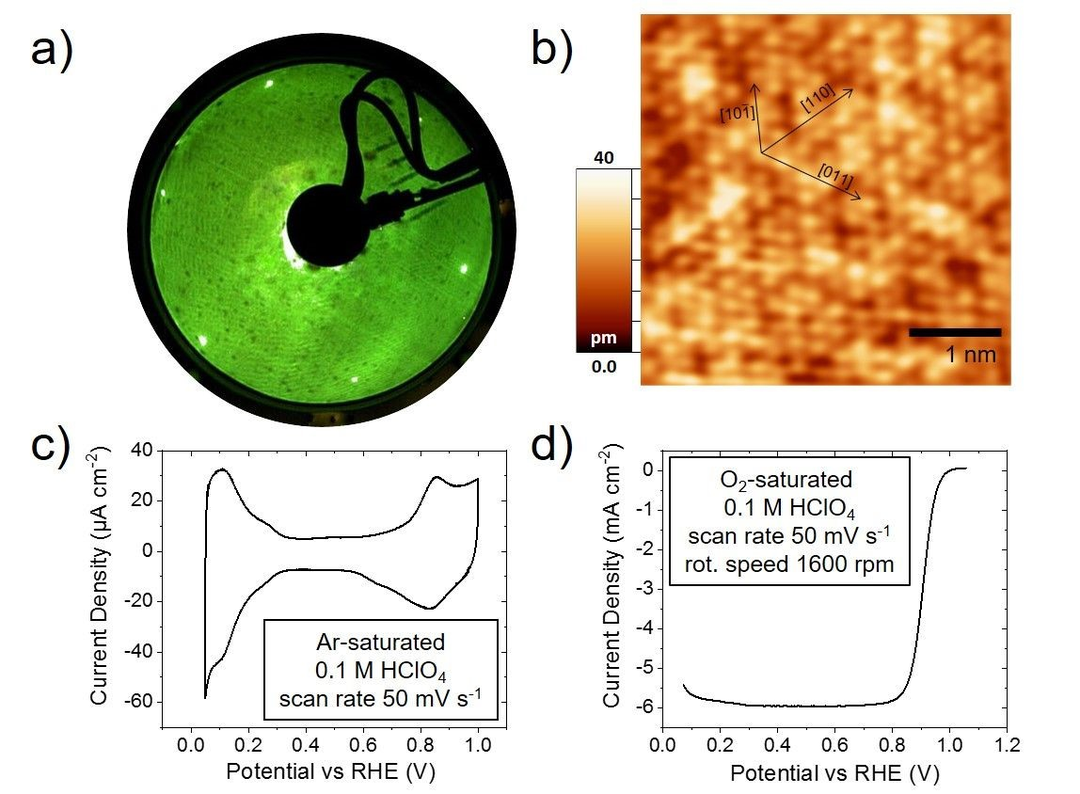
图1 新制备的Pt3Ni(111)表面科学与电化学研究。a)一次电子能量为73 eV时的LEED图像。b)确定(111)方向的高分辨率LT-STM图像。c)在Ar饱和0.1 M HClO4中的CV,扫描速率为50 mV s-1。d) 记录在O2饱和的0.1 M HClO4中以1600 rpm阳极扫描的极化曲线,扫描速率为50 mV s-1。
为了确定Pt3Ni增强ORR性能的活性位点位置,作者进行了n-EC-STM测量。图2a描述了横跨(111)平面的n-EC-STM测量。在扫描过程中,样品电位可以改变,使电极-电解质界面上的反应能够发生(反应“on”)或阻碍(反应“off”),而样品电位会在0 mVPt(“off”)和-100 mVPt(ORR“on”)之间交替。
当反应“on”时,与没有反应时相比,可以观察到噪声水平有相当大的增加。噪声表现为STM信号中不规则出现的高强度值,可以在STM图像中看到白点,在相应的剖面中看到峰值(线图扫描如图2b所示)。在(111)平台上,噪声均匀分布,表明表面由相同的活性位点组成。在台阶位边缘区域,噪声的程度似乎与平台位相同。通过分析图2b中的反应“on”和反应“off”的跨台阶直线扫描(扫描方向的高度剖面),发现在台阶位和平台位的噪声水平(尖刺的高度和密度)是相似的。
按照之前报道的量化方法,直方图采用高斯分布拟合,提取半峰全宽(FWHM)来量化噪声水平。FWHM与噪声水平的相关性如下:在噪声水平较低的情况下,信号失真不大,对应的直方图呈窄分布,FWHM较小。类似地,在高噪声水平下,信号经历明显的波动,并显示具有大FWHM的宽直方图。
图2c-d分别给出台阶位和平台位的直方图,并对反应“off”(黑色)和“on”(绿色)进行比较。如果没有反应发生,则直方图较窄,这表明噪声水平较低。相反,当ORR正在进行时,直方图变宽,对应于高噪声水平。在反应条件下,台阶位和平台位的噪声水平没有显著差异,这表明这些位点同样活跃。
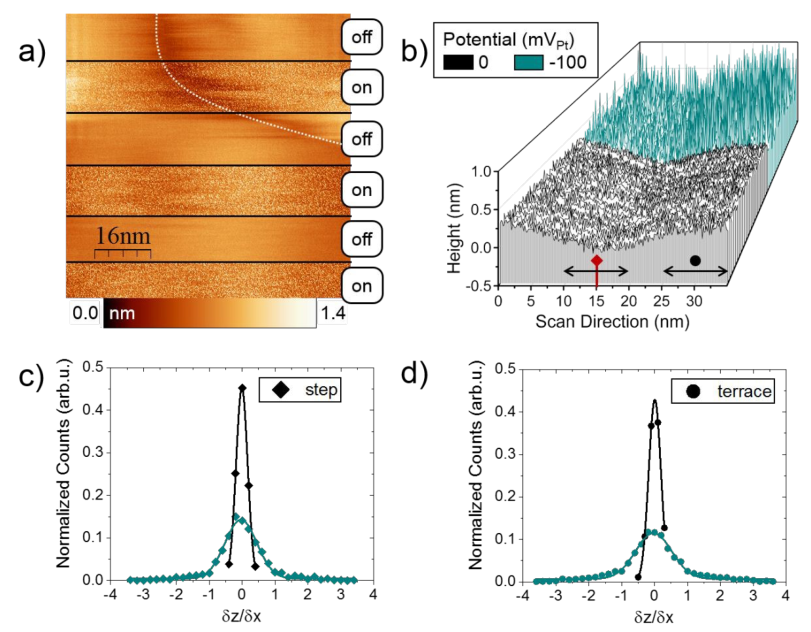
图2 a) Pt 3ni(111)平面表面的n-EC-STM测量。b)直线扫描过(a)中的面边缘。c,d)台阶位和平台位的信号导数直方图。
为了进一步将活性位点的性质与应用电位联系起来,进行了n-EC-STM测试。在图3中,可以看到(111)曲面的两个规则台阶边缘。在图像的第一部分,反应“off”,而第二部分,记录在-50 mVPt,标志着反应的开始。接下来,电位逐步降低到-200 mVPt,这相当于增加ORR电流。最后将反应反应“off”,以确认噪声的“可逆性”。
可以观察到,在图3a中,随着“过电位”的增加,噪声水平的总体范围更强。噪声水平的程度与活动有关,在台阶位和平台位均类似,这在图3b中对应的行扫描中可以更容易观察到。另外,在图3c-d的直方图中给出了定量确认。对于每一种应用电位,台阶位和平面位的FWHMs均具有可比性。此外,整体噪声水平表现位明显的随着电位的降低而增加。因为增加的反应速率导致隧道介质中更强的波动,因此产生了更多的信号噪声。
从记录的噪声的均匀外观推断出,Pt3Ni(111)表面仅由活性位点组成。(111)面在低折射率的Pt3Ni (hkl)表面中是最活跃的,这一观察在之前已经有过,并用于合成高性能的八面体Pt3Ni粒子。这可以通过比较Pt3Ni(111)和Pt(111)上*OH的结合能来解释。计算结果表明,最佳ORR催化剂与*OH的结合能(~0.10~0.15 eV)比Pt(111)更弱。
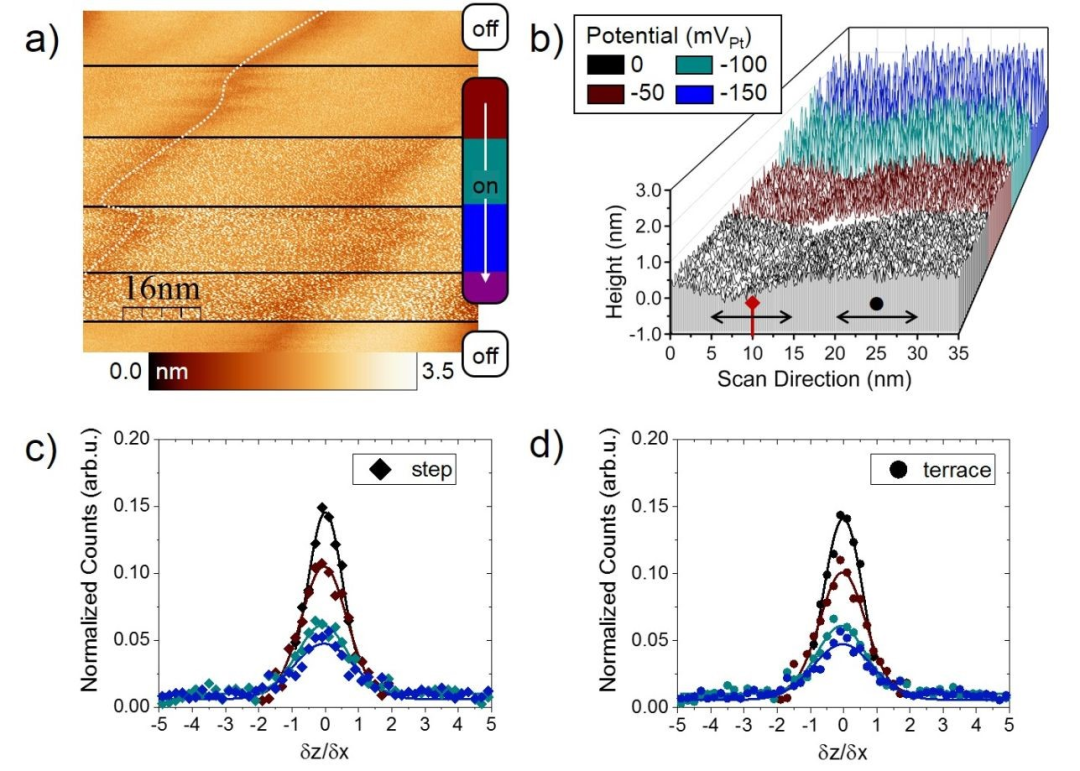
图3 a) Pt3Ni(111)表面两个规则台阶位边缘的n-EC-STM测量。b)瀑布图,包括扫描方向的高剖面,跨越(a)标记的台阶。根据数据,绘制了(c)台阶和(d)平台位点的直方图。
在本文研究中,得益于在高分辨率和原位的条件下进行的研究,可以观察到与台阶位点相比,平台位点活性更低。不幸的是,n-EC-STM并无法确定台阶位边缘的方向。为了弥补这一技术的固有缺陷,使用结构敏感计算电催化模型对Pt3Ni(111)和类似合金上活性位点的性质进行更全面的评估(图4)。
图4a提供了将合金效果合并到CN*(压缩应变和拉伸应变简单地纳入广义配数)的简单方法。首先,基于纯Pt位点的*OH结合能和CN*,建立了一个平接近化学精度的平均和最大绝对误差校准曲线(MAE=0.03 eV, MAX=0.10 eV)。随后,该曲线被用来估算Pt3M合金(M = Cu,Ni,Co,Ti)上位点的CN*。从这些得到的值与从类似纯Pt位点获得的值之间的差异,可以计算出M邻域在Pt邻域方面的等价性。
结果表明Cu,Ni,Co和Ti原子分别等价于1.70±0.12,1.59±0.25,1.48±0.05,1.42±0.17个Pt原子。这些值可用于评估Pt3M合金各种几何位点上的CN*。一旦知道了合金位的CN*,就可以用图4b中的配位-活性图来预测其ORR活性,该图将ORR极限电位(DU)与活性位的广义参量联系起来。
此外,图4c的上半部分显示了配体和应变效应如何使Pt3Ni(111)在分析的Pt3M(111)合金中具有最大的活性增强。最后,将结果与图4c底部内嵌的实验数据进行比较。在作者得到的数据集中,得到活性增强最高的催化剂是含0.5 ML Cu的PtCu NSAs(活性增强率AEexp=2.08),这相当于计算值的72%。
作者将这个数字作为所有其他电极的比例因子,包括纯Pt,Pt3Ni,Pt3Co和PtCu NSAs的数据,发现平均绝对误差只有0.12,证实了理论和实验之间的一致性表明。图4很好的解释了适当的应变和配体效应的相互作用可以推动Pt基材料向ORR活性最优方向发展,并为具有结构和成分预测性的电催化活性位点的计算设计开辟了道路。
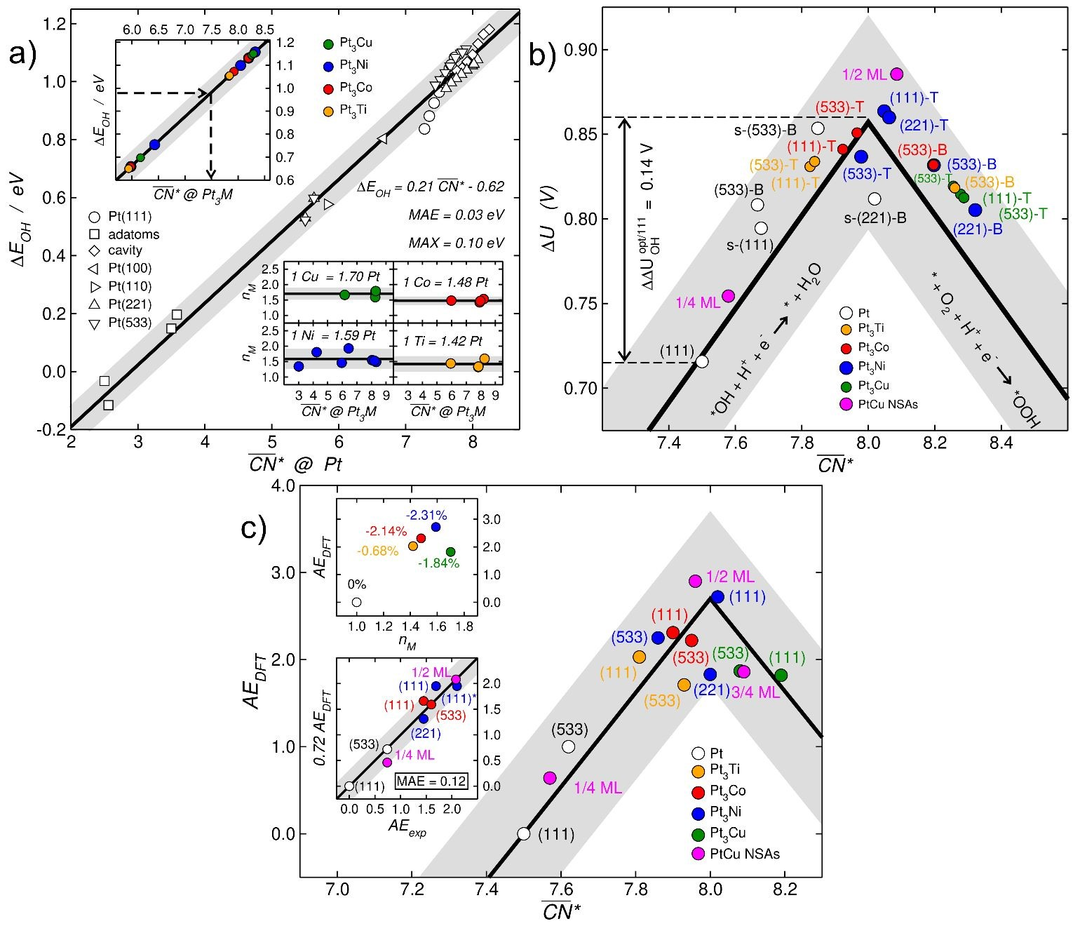
图4 Pt基电催化剂ORR的计算模型。a)DFT计算了*OH在多种纯Pt位点上的结合能。给出了线性拟合及其相关的均值和最大绝对误差(MAE, MAX)。b)纯铂和铂合金区域的配位活度图。c)对Pt(111)的活性增强(AEDFT)是电极玻尔兹曼加权广义配数的函数。
05
成果启示
该工作将n-EC-STM应用于Pt3Ni(111)单晶电极上,以确定其在酸中的ORR活性位点。结果表明,与纯Pt相比,Pt3ni(111)的基平面上的活性位点主要位于靠近台阶位的凹位点上。此外,作者设计了合金敏感的广义配位数,并创建了特定位点和特定材料的ORR活动图。通过应变和/或配体效应,能够评估Co,Ni和Cu增加了多少Pt表面位点的广义配位。该工作中实验和计算工具的相互作用,可以概述许多其他合金和电催化反应的活性位点的结构和组成。
审核编辑:刘清
-
应变效应2018-02-22 6400
-
霍尔效应和霍尔效应法测量螺线管线圈内的磁场2009-11-03 1970
-
使用大数据进行社交网络中的从众效应和权威效应影响分析2019-10-30 1506
-
内光电效应和外光电效应的区别2020-08-04 31950
-
热电偶、热电效应和热电效应原理2021-10-29 10544
-
阐明Pt单原子催化剂的轴向配体效应对碱性析氢反应的影响2023-02-02 3245
-
MOS管的米勒效应:如何平衡抑制米勒效应和抑制EMI风险的关系2023-04-17 10219
-
介绍一种扁线电机趋肤效应和邻近效应的新型解决方案2023-07-18 3602
-
电阻应变计的温度效应2023-08-24 2562
-
半导体应变片的应变效应?2024-05-16 3912
-
电流的磁效应和电磁感应的区别2024-07-18 18264
-
使用 TI 的霍尔效应和线性 3D 霍尔效应传感器替代簧片开关应用说明2024-09-10 738
-
电光效应之普克尔效应和克尔效应2024-12-02 3243
-
霍尔效应和量子霍尔效应的原理与机制2025-01-07 3492
全部0条评论

快来发表一下你的评论吧 !
