

设计一款新的二乙二醇二甲醚(DME)基电解液
描述
【研究背景】
电解液溶剂化结构化学最近引起了电池领域研究者的极大关注,其主要原因是传统认为的通过电解液在电极表面分解形成的固体电解质界面膜(SEI)并非稳定电极及电池性能的唯一主导因素。这一观点虽然已经通过循环的石墨、合金、金属负极以及多数正极在“电解液交换实验”中得到证明,但是并不能验证SEI膜在某些方面可能的积极作用。
因此,如何区分电解液溶剂化结构及其界面模型与SEI对电极及电池性能的影响仍具挑战。尤其将成膜添加剂引入电解液中时,该问题变得更加复杂,因为添加剂有可能形成SEI,亦有可能改变电解液溶剂化结构,然其各自的作用极难以辨别和区分。
针对上述问题,本研究以锑(Sb)负极为例,设计了一款新的二乙二醇二甲醚(DME)基电解液,详细阐述了溶剂化结构及其衍生的界面模型和SEI对电极性能的影响,尤其对添加剂在电解液体相、电极界面以及SEI成膜过程中的作用进行了详尽解析。
近期,中科院长春应化所明军,兰州大学张俊丽,以及韩国汉阳大学Yang-Kook Sun阐明了电解液组分(溶剂、锂盐、浓度、添加剂等)从溶剂化结构、界面模型到 SEI 成膜过程中的分子、离子行为以及各自产生的作用,发现了独特的溶剂化结构衍生的界面模型和SEI均是稳定Sb负极的关键。
其中,添加剂(二氟草酸硼酸锂,LiDFOB)不但能削弱Li+-DME之间的相互作用,而且参与形成的特定的SEI膜能够有效削弱电极给电子的能力,进而提高电解液及电极稳定性。
【内容表述】
1.研究主旨
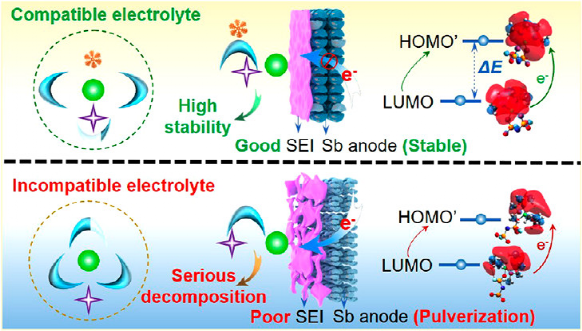
图1. 溶剂化结构衍生的界面模型和SEI对电极的影响。
本研究以Sb负极例,详细研究了溶剂化结构衍生的界面模型和SEI对电极稳定性的影响(图1)。
2.电化学性能表征
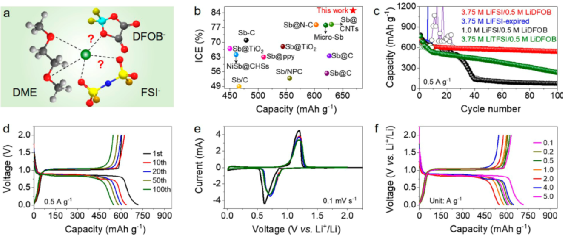
图2. 微米Sb负极电化学性能。
本研究设计了一款以LiFSI锂盐,DME溶剂,LiDFOB添加剂的电解液(即3.75 M LiFSI/0.5 M LiDFOB in DME),用以稳定微米Sb负极,在该电解液中表现出656 mAh/g的容量及87.5%的首效,优于之前报道的工作(图2a, b)。0.5 A/g电流密度下,100次循环后容量保持率88.6%,表现出优异的电化学性能。
相比之下,在没有LiDFOB添加剂的3.75 M LiFSI in DME电解液中,微米Sb负极在几个循环后就无法再正常充电。将电解液浓度降低至1.0 M(即1.0 M LiFSI/0.5 M LiDFOB in DME)时,微米Sb负极容量急剧下降,约40次循环后容量仅剩~130 mAh/g。
此外,使用LiTFSI锂盐(3.75 M LiTFSI/0.5 M LiDFOB in DME),Sb负极容量也大幅衰减,容量由初始的626 mAh/g经100次循环后降至229 mAh/g。以上结果表明,电解液的组分(如锂盐、溶剂、浓度、添加剂)均会影响微米Sb负极性能。
3.电极及SEI膜的表征
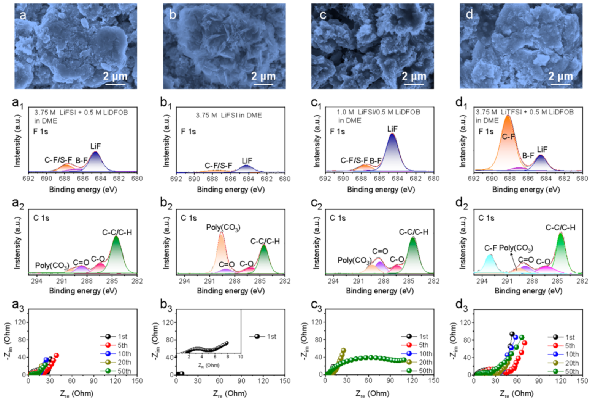
图3. 电极及SEI膜的表征。
为确定不同电解液对微米Sb负极影响的原因,研究者对循环后的微米Sb负极进行了相关表征(图3)。
结果表明,(1)3.75 M LiFSI/0.5 M LiDFOB in DME电解液中循环后的微米Sb负极表面的SEI比较光滑,且电极粉化程度最小,证明了高浓度和添加剂是维持微米Sb负极稳定的必要条件;(2)从F1s和C1s谱图可以看出,3.75 M LiFSI/0.5 M LiDFOB in DME电解液中,微米Sb负极表面形成了一层以LiF为主导(LiF/含F有机化合物,2.74)且C含量少的SEI,表明在该电解液中,DFOB-的分解量有限,生成的SEI稳定,能有效抑制溶剂的分解;(3)3.75 M LiFSI/0.5 M LiDFOB in DME电解液中循环后微米Sb负极的电荷转移电阻仅为4.11 Ω,这有利于Li+在电极中的扩散。
4.SEI膜作用分析
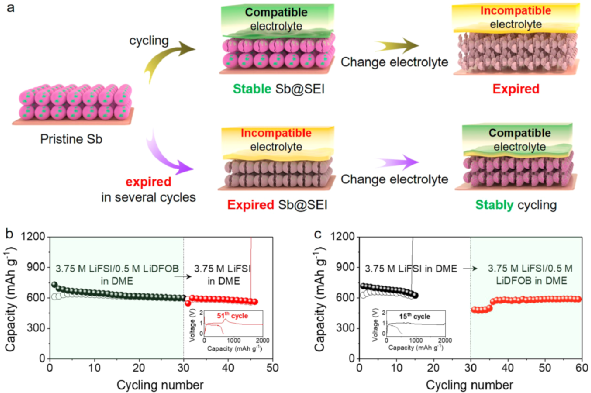
图4. 电解液交换实验。
尽管图3中对SEI的表征结果看似可以解释图2c中微米Sb负极不同的电化学性能,但对SEI的分析仍不能全面地解释电解液交换实验中电极性能的不同。具体的交换实验如图4a和b所示。首先,让微米Sb负极在兼容电解液(即3.75 M LiFSI/0.5 M LiDFOB in DME)中循环30次,以此在电极表面形成稳定的SEI(Sb@SEI);然后,拆开电池,使用Sb@SEI负极重新组装电池,使用不兼容电解液(即3.75 M LiFSI in DME)继续循环。
从图4c可以发现,在不兼容电解液中,Sb@SEI负极能够维持14次循环,在第15次循环过程中失效。以上结果表明,如果电解液不能兼容微米Sb负极,即便在兼容电解液中生成的SEI可以起到保护微米Sb负极的作用,但作用有限,不能在长循环中持续稳定负极。
相反地,首先让微米Sb负极在不兼容电解液(即3.75 M LiFSI in DME)循环,直到其不能正常工作,得到在差的SEI包覆的微米Sb负极(Sb@SEI);然后,拆开电池,使用Sb@SEI负极重新组装电池,使用兼容电解液(即3.75 M/0.5 M LiDFOB LiFSI in DME)继续循环。从图4d可以观察到,Sb@SEI负极经过5次循环后可恢复初始的容量,并能维持良好的循环稳定性。以上结果表明,微米Sb负极失效的原因可能是电解液的不兼容。
5.体相电解液性质表征
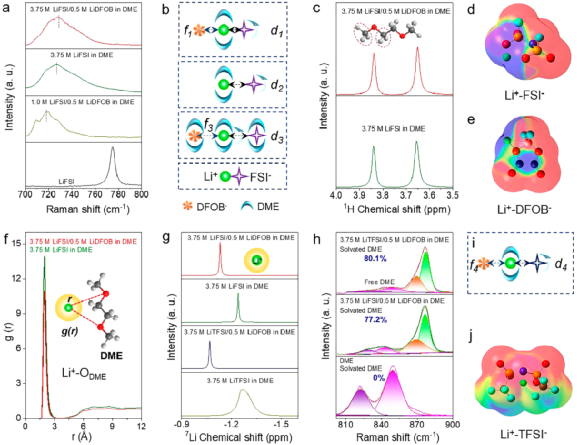
图5. 电解液表征。
因此,本研究将重点从分子层面上构连电解液组分与电池性能之间的关系。通过拉曼光谱、液相核磁共振谱等测试分析了不同电解液中的Li+、溶剂、添加剂之间的相互作用,以明晰不同电解液中Li+溶剂化结构的差异(图5)。拉曼结果表明,通过提高锂盐浓度(如将电解液浓度从1.0 M提高到3.75 M)或引入LiDFOB添加剂可以显著降低S-N-S的红移程度。
这主要是因为随着锂盐浓度的提高或添加剂的引入,参与溶剂化的DME数量逐渐不足。换句话就是,红移程度越小,Li+-FSI-的相互作用越强,即3.75 M LiFSI/0.5 M LiDFOB in DME电解液中Li+-FSI-的相互作用最强(图5a, b)。在1H核磁共振谱中,DME溶剂分子中的-CH2/-CH3基团的化学位移进一步证明了引入LiDFOB添加剂能增强Li+-FSI-的相互作用(图5c)。当LiDFOB引入3.75 M LiFSI in DME电解液中,1H核磁共振谱中对DME中-CH2/-CH3基团的屏蔽作用会增大。
这主要是因为LiDFOB也需要DME溶剂参与解离,导致溶剂化结构中DME溶剂分子数量不够,在一定程度上减弱了Li+-DME的相互作用。这一观点通过径向分布函数计算结果得到了进一步证明(图5f)。此外,通过7Li核磁共振谱进一步证实了LiDFOB添加剂对Li+溶剂化结构的影响。
如图5g所示,将LiDFOB引入3.75 M LiFSI in DME电解液中后,7Li核磁共振谱的化学位移向低场偏移表明溶剂对Li+的屏蔽作用降低,这主要是由于DME溶剂数量不足和DFOB-在Li+周围频率出现增大引起的。这些有力的证据表明,LiDFOB除了众所周知的成膜功能外,还能参与Li+溶剂化结构。
6.溶剂化结构及其界面模型
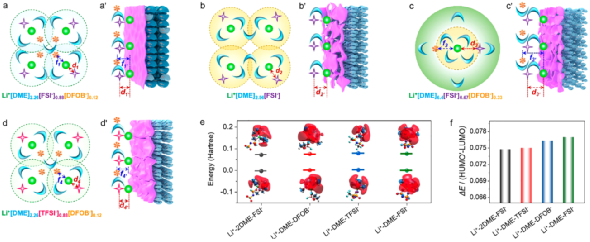
图6. Li+溶剂化结构及界面模型。
根据Li+在不同电解液中与溶剂、阴离子之间的相互作用,建立了Li+溶剂化结构及其衍生的界面模型。其中,电解液以其结构化单元Li+[solvent]x[anion]来表示。此外FSI-和Li+之间的相对距离(即d')以及DFOB-和Li+之间的相对距离(即f')用于定性描述Li+去溶剂化过程中微米Sb负极表面Li+-FSI-和Li+-DFOB-相互作用强度,距离越长,则代表相互作用越弱。
此外,用最低的未占用分子轨道(LUMO)和获得一个额外的电子时,相应轨道变为HOMO'的能量差(ΔE)来评估电解液的稳定性。其中,较小的能量差异表示轨道之间的相似性,导致电子传输所需要的能量越低,说明了Li+-solvent-anion复合物稳定性越弱,即电解液也不稳定。
在3.75 M LiFSI/0.5 M LiDFOB in DME (Li+[DME]2.26[FSI-]0.88[DFOB-]0.12)电解液中,由于DME溶剂不足,不足以将Li+、FSI-和DFOB-充分溶剂化,Li+第一溶剂化层会共享溶剂而相互重叠。由于Li+-FSI-和Li+-DFOB-的相互作用较Li+-DME强,FSI-和DFOB-会出现在第一溶剂化层中的Li+周围,削弱Li+与DME之间的相互作用,进而降低DME溶剂分子的极化。在去溶剂化过程中,可以在微米Sb负极表面形成以阴离子为主的电解液分解,有效抑制了溶剂的分解,提高了电解液及微米Sb负极的稳定性(图6a)。
此外,理论模拟得到,Li+-DME-DFOB-配合物的ΔE(0.0750 Hartte)略小于Li+-DME-FSI-配合物的(0.0770 Hartte)(图6f)。这说明,Li+-DME-DFOB-配合物更容易接收电极上的电子,并在电极表面还原形成SEI。随后,阴离子主导分解形成的SEI会削弱电极对Li+-DME-DFOB-或Li+-DME-FSI-配合物的供电子能力,提高了电解液的稳定性。基于上述对电解液稳定性的分析,成功地阐明了微米Sb负极的优异性能的原因。
相比之下,在3.75 M LiFSI in DME (Li+[DME]2.56[FSI-])电解液中,由于没有LiDFOB分散DME溶剂参与解离溶剂化,参与解离溶剂化LiFSI的DME溶剂分子数量增多,第一溶剂化层重叠的程度较有添加剂的减少。换言之,Li+-FSI-的相对距离(d2)略长于Li+[DME]2.26[FSI-]0.88[DFOB-]0.12中Li+-FSI-的相对距离(d1,d1 < d2)。
同样的,在图6b'的界面模型中Li+与FSI-之间的相对距离(d2')也相对远一些,即d2' > d1'。在去溶剂化过程中,FSI-虽然也会优先接收电极上的电子参与形成SEI,但与DFOB-衍生的SEI膜相比,FSI-诱导生成的SEI膜并不稳定,不足以有效的削弱电极的供电子能力。
这样会导致Li+-DME或Li+-2DME-FSI-配合物可以继续得到电子发生分解,造成SEI中C含量的增加,在电极表面形成富C和F有机物的SEI。以上因素共同影响,使得微米Sb负极在几次循环后就不能正常工作。在Li+[DME]9.6[FSI-]0.88[DFOB-]0.12电解液中进一步证实了LiDFOB添加剂的作用。当LiFSI浓度降低到1.0 M时,由于参与溶剂化的DME溶剂分子数量足够多,Li+溶剂化结构(d3,f3)和界面模型(d3',f3')中Li+和FSI-、DFOB-的相对距离最长(图6c,c')。
此外,Li+-2DME-FSI-配合物的ΔE值为0.0745 Hartte,小于Li+-DME-FSI-配合物的0.0770 Hartte(图6f),表明Li+[DME]9.6[FSI-]0.88[DFOB-]0.12电解液的稳定性相比高浓度电解液要差。在去溶剂化过程中,DFOB-和DME溶剂分子会很容易在电极表面得到从电极上传递的电子发生分解,在电极表面生成富B、C和F物质的SEI。
生成的SEI可以在一定程度上抑制电极的供电子能力,相对减少DME的分解,使得微米Sb负极可以维持基本的循环。然而,在Li+[DME]9.6[FSI-]0.88[DFOB-]0.12电解液中,由于DME溶剂能充分解离、溶剂化锂盐,DME溶剂分子被Li+的极化程度较高,会不能有效地抑制循环过程中电解液的分解,在电极表面生成富含大量有机物的SEI,导致电极阻抗逐渐变大。
以上结果进一步表明提高锂盐浓度和引入LiDFOB添加剂,可以提高电解液稳定性,改善电极性能。此外,选择合适的阴离子对维持电解液的稳定性也很重要。如图6d,d'所示,在Li+[DME]2.26[TFSI-]0.88[DFOB-]0.12电解液中,Li+溶剂化结构和界面模型中Li+-TFSI-(d4,d4')的相对距离与Li+[DME]2.26[FSI-]0.88[DFOB-]0.12电解液中的相对距离要远一些,而Li+-DFOB-(f4,f4')的相对距离则相当。
在去溶剂化过程中,由于Li+-DME-DFOB-配合物较低的ΔE值,使得DFOB-更容易在电极表面发生分解,并形成富B和F物质的SEI。虽然生成的SEI能够削弱电极的供电子能力,但由于Li+-DME-TFSI-配合物的ΔE值(0.0760 Hartte)低于Li+-DME-FSI-配合物的,且Li+对DME溶剂分子的极化程度相对较大,造成电解液的分解,最终导致电池容量的衰减。
以上研究结果表明,Li+溶剂化结构及其界面模型与SEI能有效地改善微米Sb负极电化学性能。其中SEI可以有效地降低电极的给电子能力,减少电解液的分解,进而提高电解液的稳定性。
7.锂离子全电池应用
为了进一步验证所设计电解液的优势,组装了以微米Sb为负极,LiFePO4做正极,3.75 M LiFSI/0.5 M LiDFOB in DME做电解液液的锂离子全电池,如图7所示。该电池在0.5C(1C = 150 mA/g)电流密度下表现出了良好的循环性能,经100次循环,容量保持在78.4 mAh/g,容量保持率为54%。
此外,在0.1、0.2、0.5、1、2和5C电流密度下,容量分别为155、151、141和126 mAh/g,表明了全电池具有优异的倍率性能。上述结果充分证明了微米Sb负极在3.75 M LiFSI/0.5 M LiDFOB in DME电解液中的高稳定性和可逆性。
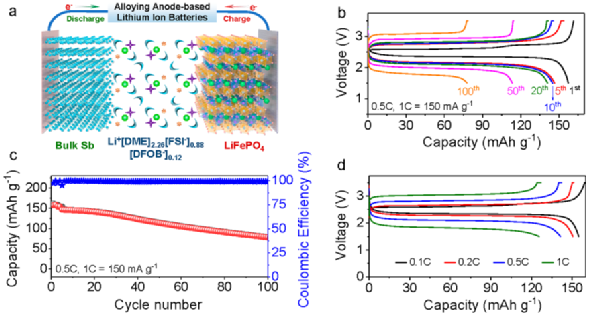
图7. 全电池性能。
【结论】
该工作以微米Sb负极为研究对象,设计了一款新的基于DME的电解液,以认识溶剂化结构衍生的界面模型和SEI对电极性能的决定性影响。研究发现,锂盐浓度、种类和添加剂对于特定SEI和界面模型的形成都至关重要,最终影响电极的循环稳定性。尤其,该工作从溶剂化结构、界面模型到最终形成的SEI的动态演化过程全面阐述了电解液组分尤其添加剂的作用。该研究为通过调节溶剂化结构和SEI的协同方法设计电解液提供了指导。
审核编辑:刘清
-
电解液——锂电池的‘血液’2018-08-07 5926
-
锂离子电池材料之电解液(详细篇)2009-11-03 20020
-
直接二甲醚燃料电池2011-02-24 1135
-
合成气直接合成二甲醚的动态建模2018-01-13 1007
-
车载乙二醇防冻液储存箱液位监控仪表的应用2018-12-19 3063
-
锂电池电解液溶剂基础入门知识总结2021-03-26 4658
-
一种有效的开发高性能电解液的解决方案2022-09-05 2861
-
工业用乙二醇紫外透光率的测定方案(紫外分光法)2022-10-13 7102
-
FEC+DME电解液新配方2022-11-08 5719
-
局域高浓电解液的形成及抗氧化分析2022-11-22 2202
-
二甲醚球罐液位计应用案例2023-06-15 1148
-
用差示扫描量热仪(DSC)聚对苯二甲酸乙二醇酯(PET) DSC测试2023-01-31 1863
全部0条评论

快来发表一下你的评论吧 !
