

经典熵态调节提升正极材料可逆容量
描述
01
研究背景
目前人们对于高容量正极的需求愈发迫切,高电压可提升正极材料比容量,但是传统的过渡金属氧化物会出现TM和Li的迁移。导致材料结构、对称性及物相改变。因此,构建高容量正极材料的关键之一是防止阳离子的持续重排和迁移。
研究发现,LixTMyO2的对称性变化是由不同状态下的位点能量转移所驱动的, LixTMyO2型的热力学稳定相一般随Li含量的变化而变化。对于大多数具有阳离子有序的三维TM基LixTMyO2,即低熵态相,阳离子无序的出现在高容量运行(>200 mA h g−1)时是热力学不可避免的,如图1所示。
最近,文献报道了使用无序岩盐(DRX)正极并施加1.5 V的低截止电压时,放电容量高于300 mA h g−1(在较低的比电流下,如<20 mA g−1,且循环次数非常有限)。在这些材料中,所有的阳离子都随机分布在4a位点上,形成了比阳离子有序的LixTMyO2更高的熵态。
虽然通过特殊策略可以获得具有快速Li+扩散动力学的DRX,但在这些高熵DRX相中Li+通过渗流路径的扩散受到Li+浓度和局部条件的限制,实际应用中难度较大。
02
成果简介
近日,哈工大(深圳)甄良教授、徐成彦教授与南京大学王鹏团队在Nature Communications上发表题为“A medium-entropy transition metal oxide cathode for high-capacity lithium metal batteries”的研究论文。本文报道了一种缺陷态正极材料Li1.46Ni0.32Mn1.2O4-x(0 < x < 4),在首次充电后出现中等熵态的局部阳离子无序尖晶石相,并提出了Li+从八面体稳点向尖晶石结构位点的转移过程以及Mn元素的+3/+4电荷补偿机制。
该熵态调节机制的提出为高容量正极材料设计提供了新的思路。
03
研究亮点
(1) 以Li1.46Ni0.32Mn1.2O4-x (0 < x < 4)材料为研究对象,首次提出了熵混乱度变化的概念,表明其充电脱锂后会出现局部阳离子无序的尖晶石相,提升材料循环性能。
(2) 通过理论计算和理化标准证明了Li+从八面体稳点向尖晶石结构位点的转移过程,提出了以及Mn元素的+3/+4电荷补偿机制。
04
图文导读
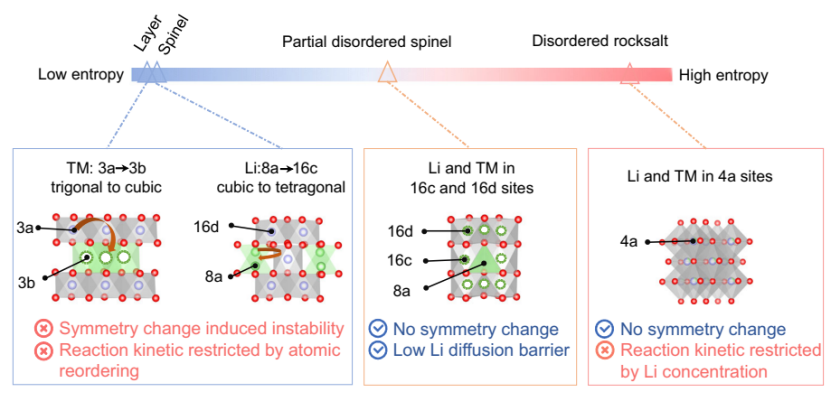
图1 在不同的LixTMyO2相中,充放电过程中阳离子的重新排序和对称性变化的示意图. 3a、16c、16d和4a八面体位用灰色八面体表示,3b八面体位和8a四面体位分别用绿色八面体和四面体表示. 过渡金属离子、锂离子和氧离子分别用紫色、绿色和红色的球表示,空位的可能占用率用绿色的虚线表示.
缺陷Li1.46Ni0.32Mn1.2O4-x(0 < x < 4)材料制备是在富锂层状氧化物中通过质子交换和操纵阳离子重排实现的(CD-LNMO)。如图2a所示,尽管CD-LNMO中的Li:TM比值位于LLO和LNMO之间,CD-LNMO的电化学行为与之前报道的LLO/LNMO复合材料不同。
通过同步辐射SXRD研究了CD-LNMO在第一个循环过程中阳离子有序的演变过程,得到了LLO和LNMO的XRD谱图。如图2b所示,合成的CD-LNMO是层状/尖晶石复合材料,脱锂后的样品(充电到4.8 V)的峰形向无杂质相的纯尖晶石相转移。EXAFS结果表明,合成的CD-LNMO的Mn/Ni-O键的共存(图2c)表明,这意味着在缺陷产生后,TM离子的局域构型发生了改变。
考虑到Li在x射线测量下散射因子较低,因此补充了进行中子衍射测量,以表征Li离子的占位情况(图2d, e)。
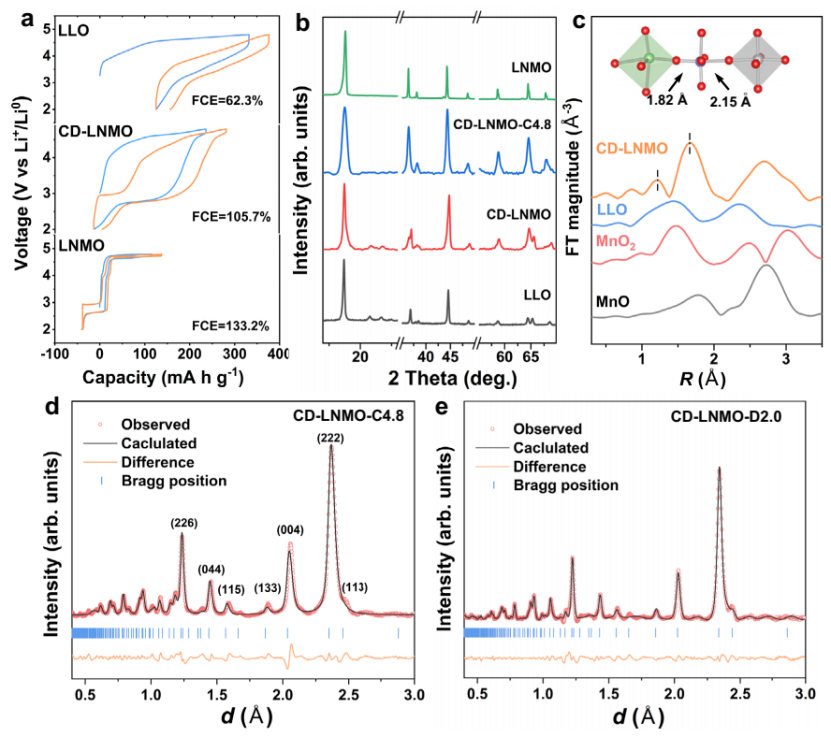
图2 不同锂过渡金属氧化物材料的电化学和结构表征. (a) 三种材料室温下的充放电曲线. (b) CD-LNMO和充电态CD-LNMO的同步辐射XRD图. (c)LLO和CD-LNMO的K3加权傅里叶变换Mn K边EXAFS谱. CD-LNMO充电态4.8 V (d)和放电态2V (e)的TOF中子衍射谱.
CD-LNMO的TEM和HAADF-STEM图像显示两个不同对比度区域共存(图3a-c)。FFT变换后的HAADF-STEM图像在弥散的低对比度区域显示三维尖骨石有序结构,而高对比度区域可能保留有部分TM离子无序的层状相,表明CD-LNMO中存在阳离子无序结构。在充电过程中,CD-LNMO中Li+的脱出促进了TM离子的3D排序(图3d)。
如图3e-g所示,高分辨HAADF-STEM信号显示了TM离子在尖晶石型骨架的16c和16d位点的占位情况,证实了脱锂后尖晶石结构的部分无序。
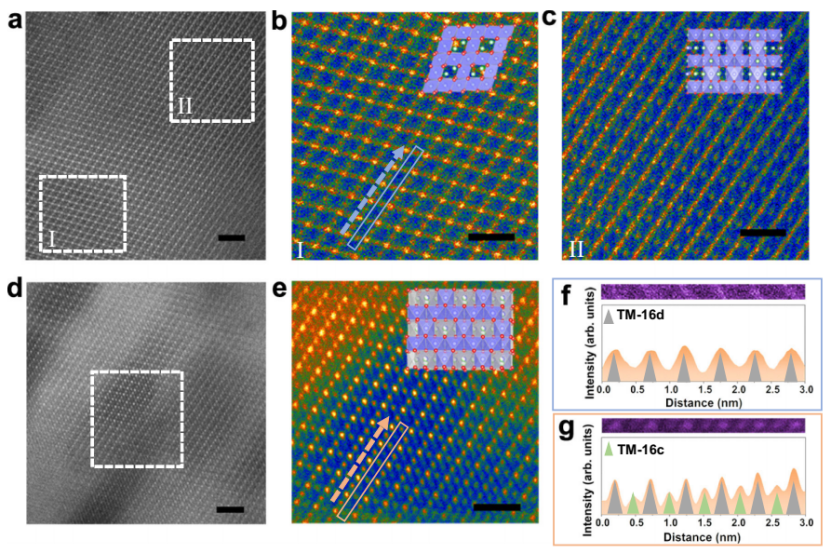
图3 非原位Li1.46Ni0.32Mn1.2O4-x电极的非原位STEM. (a) CD-LNMO的HADDF-STEM图. I区(b)和II区(c)的FFT变换谱. 经过10次活化循环的NVPF-H和NVPF-U正极的SEM 图. (d-e) 满充电态的CD-LNMO粉末HADDF-STEM图. (f)和(g)对应b和e图中框线部分放大的HADDF-STEM图.
进一步探究了电化学性能,电池在2.0-4.8 V, 25±5°C内循环。如图4a所示,CD-LNMO表现为中等水平的电压下降,每循环约1.6 mV,与LNMO相当,但远小于LLO(约9.4 mV /循环)。循环过程中,CD-LNMO、LLO和LNMO的放电容量在初始循环中逐渐增加(图4b-e),这可能与电极的逐渐活化有关。
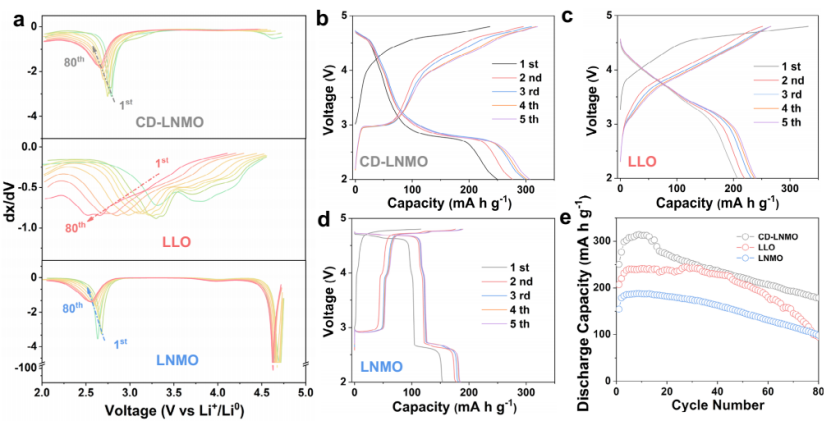
图4 (a) CD-LNMO、LLO和LNMO第1 ~ 80循环的微分容量曲线. CD-LNMO (b),LLO (c),和)LNMO(d前五圈充放电曲线. (e)三种材料的循环稳定性(2-4.8 V).
为了可视化地从部分阳离子无序中追踪到的氧化还原中心的移动,作者进行了循环伏安(CV)测试,如图5a-c所示。CD-LNMO、CD-LNMO- h和LNMO的CV曲线分别在4.7/4.6 V和3.0/2.7 V处有两个明显的阳离子氧化还原峰,分别对应于Ni2+/Ni4+和Mn3+/Mn4+的氧化还原反应。受益于快速沿着0-TM扩散路径下贫TM配位结构以及尖晶石结构框架,CD-LNMO电池展现出优异的电化学性能(图5 d)。
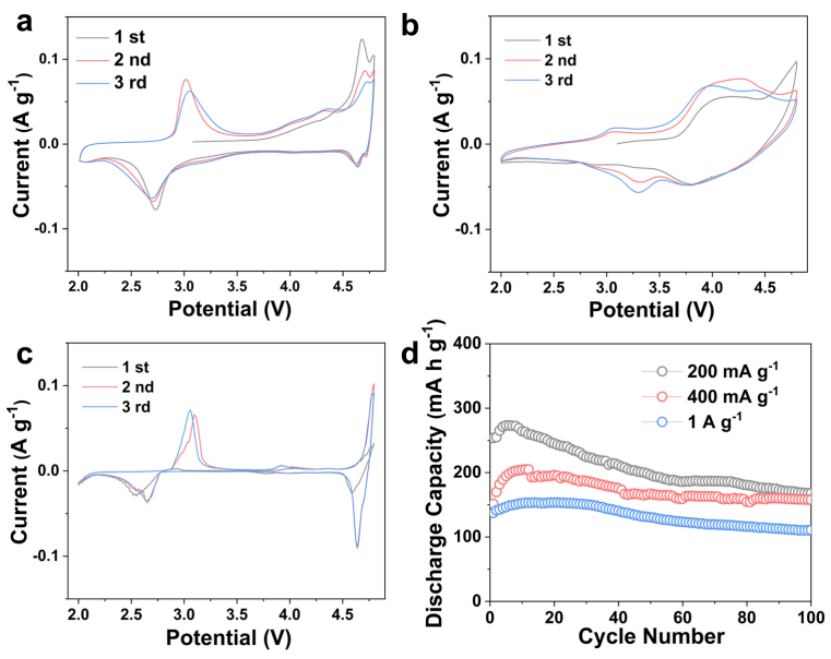
图5 Li||CD-LNMO (a)、Li||LLO (b)和Li||LNMO (c)扣式半电池的前三个循环伏安曲线. (d) Li||CD-LNMO电池在不同倍率下的循环稳定性.
采用原位k边-XANES测量方法,研究了CD-LNMO中Mn和Ni在初始循环过程中的氧化态变化。合成的CD-LNMO中Mn和Ni的吸收边与MnO2和NiO的吸收边接近,分别表明Mn和Ni的价态接近+4和+2。如图6b所示,4.8 V充电样品的mRIXS测试结果表明完全脱锂态CD-LNMO晶格中存在大量高价氧基团。
进一步分析局域结构,非原位Mn和Ni k边EXAFS显示了在初始循环中Mn-O键的演化(图6c, d)。充电后,较短的Mn-O键略有拉伸,而较长的Mn-O键保持不变(图6c)。MnO键在放电过程中进一步拉伸,很可能是由Mn离子的还原引起的,如图6d所示。
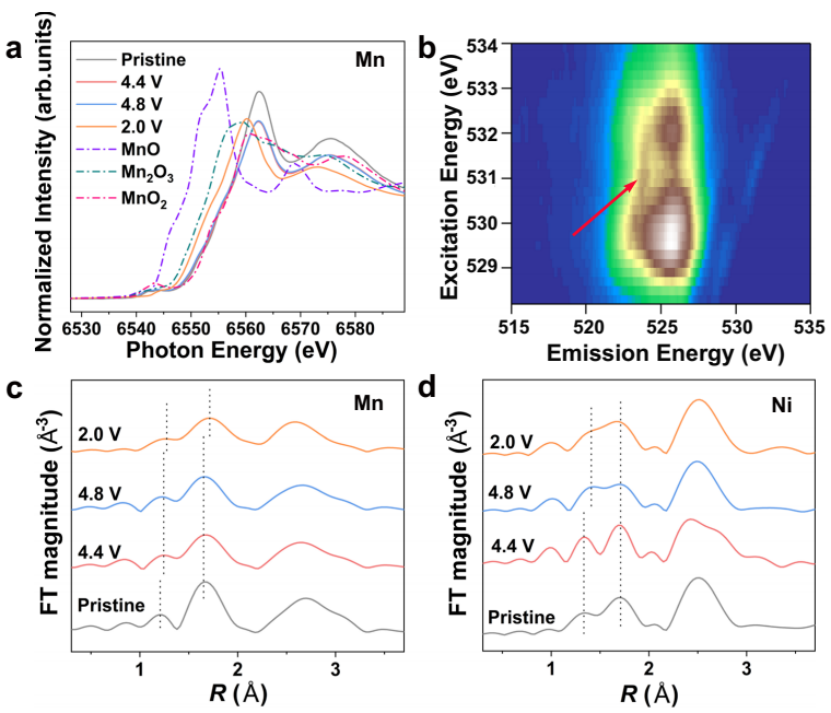
图6 全电池性能. (a) 首次循环中,在不同电压下CD-LNMO电极归一化原位Mn k边XANES光谱. (b) 4.8 V充电态O的K边MRIXS谱. 不同电压下CD-LNMO电极的 (c) Mn k边EXAFS谱和 (d) Ni K边EXAFS谱的k3加权傅里叶变换.
为了阐明CD-LNMO的阳离子无序性与电化学性能之间的关系,作者通过DFT计算来研究CD-LNMO的晶体和电子结构。如图7a所示,作者发现化学计量比的LNMO(标记为CDLNMO-1)中的阳离子混排在热力学上难以形成。通过比较16c八面体位(Lioct)和8a四面体位(Litet)的能量,探讨了阳离子无序对锂离子存储机制的影响。
如图7b所示,Litet的位点能量为0.373 eV,低于LNMO中的Lioct,这与之前的报道一致,表明Li离子优先进入四面体位置。如图7c所示,计算得到的电子定位函数显示了O2、O3和O4上的O-2p孤对轨道(lO2p),意味着这些氧离子上的阴离子反应具有更高的能级。
图7d所示,在首次充电过程中,阳离子氧化主导了电荷补偿,对CD-LNMO中lO2p的观察显示,O2−/O−的能级高于Ni2+/Ni4+,表明至少部分O2−离子位于缺乏TM的尖晶石结构中。这使得在阳离子和阴离子氧化还原过程中都可以进行可逆循环,Li离子在八面体位的穿梭不仅提高了容量,而且有效地避免了高容量时的对称性变化。
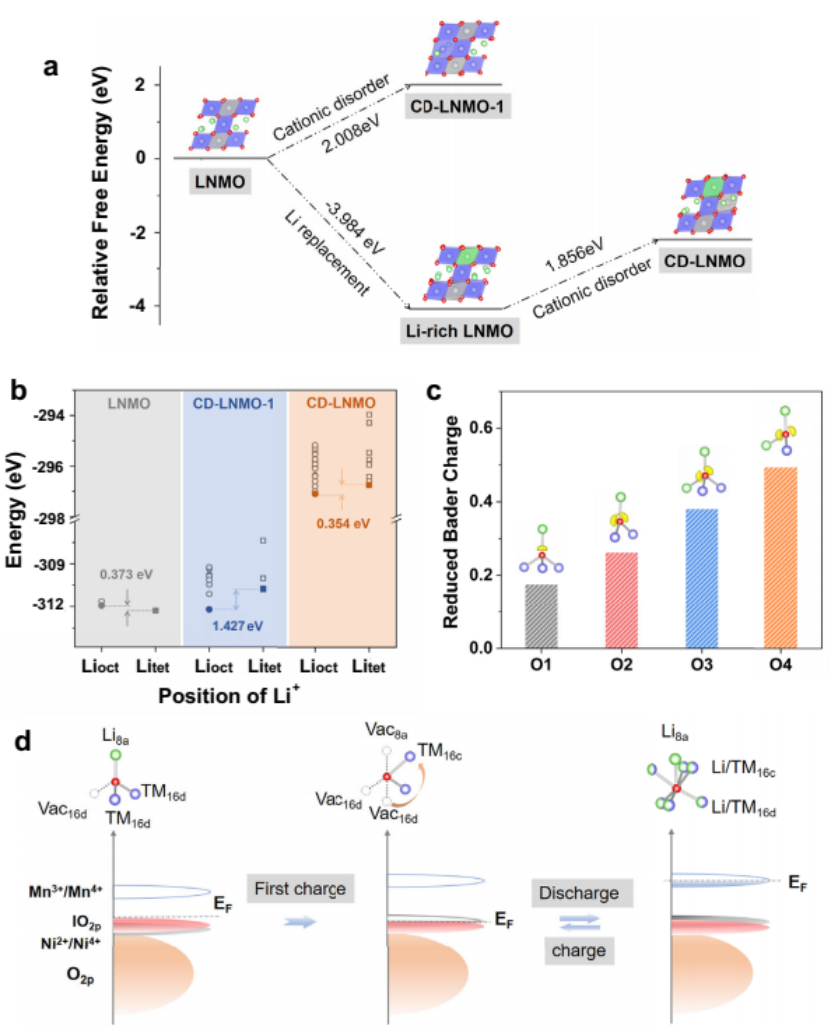
图7 (a) 计算LNMO上结构演化的自由能图. (b) LNMO, CD-LNMO-1和CDLNMO中八面体位点和四面体位点之间的能量差. (c) 完全放电CD-LNMO在不同条件下氧离子的Bader电荷减少量. (d) 循环时的定域条件和相应的电子结构演化示意图.
05
总结和展望
由于LixTMyO2正极的实际容量和理论容量之间存在很大差距,高电压时LixTMyO2的内在对称性变化是需要克服的关键之一。作者证明了一个合适的熵水平可以有效地抑制不利的对称性变化,通过3D锂离子扩散路径实现高容量。这些结果表明,熵态可以有效地调节正极材料的晶体结构和电子结构。本文提出的熵操纵策略是克服Li离子可逆嵌入脱出过程中热力学驱动的原子无序占位演化的潜在方向,而阳离子无序尖晶石结构为探索中熵化合物开辟了广阔的空间。
06
文献链接
A medium-entropy transition metal oxide cathode for high-capacity lithium metal batteries. (Nat. Commun.2022, DOI:10.1038/s41467-022-33927-0)
原文链接:
https://www.nature.com/articles/s41467-022-33927-0
审核编辑:刘清
-
有机化合物可作为锂离子电池正极材料2015-11-17 0
-
锂离子电池的最新正极材料:掺锰铌酸锂?2016-01-19 0
-
锰酸锂正极材料---理想的动力电池材料2009-10-29 1153
-
锂离子电池正极材料LiNiVO4的容量衰减原因分析2010-01-26 2619
-
钠离子电池正极材料怎样实现可逆氧变价的结构2019-02-10 1850
-
新富镍三元正极材料将有望大大降低高性能富镍三元材料的生产门槛2019-02-15 842
-
南开大学开发出一种具有超高容量的锂离子电池有机正极材料2019-05-17 1058
-
富镍三元正极材料的改性策略2020-11-02 8748
-
锂过量的正极材料中可逆的Mn2+/Mn4+双氧化还原2020-12-25 736
-
钠离子电池正极可逆氧变价机理概述2020-12-25 931
-
讲解不同正极材料锂电池容量特性2021-03-26 4248
-
调节自旋电子结构来改善富锂材料的电压滞后2023-03-09 1314
-
开发高性能高镍正极材料的多功能掺杂策略2023-08-04 1976
-
锂锰电池的正极材料是由什么组成的?锂锰电池正极材料的优点2023-11-10 796
-
高负载质量下MnO2正极材料容量衰减问题的解决方案2024-10-24 305
全部0条评论

快来发表一下你的评论吧 !
