

具有不同配位环境的Pt单原子层的可控制备研究
描述
全文速览
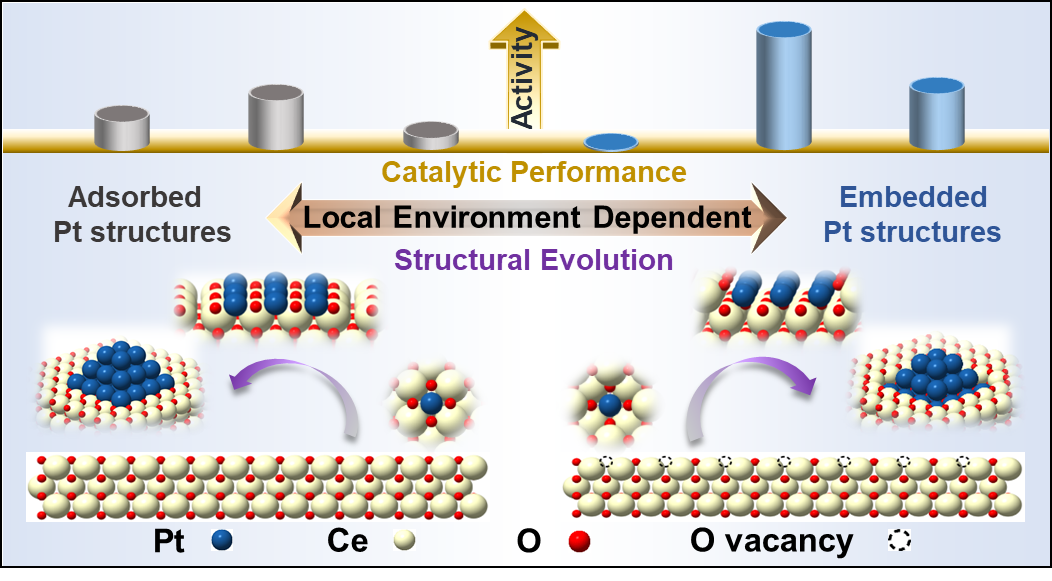
金属位点的局部配位结构决定了负载型金属催化剂的性能。在本工作中,作者利用表面缺陷富集策略,在CeO2-Al2O3载体上以铂单原子(Pt1)为前体物成功制备了具有100%金属分散和局部配位环境(嵌入与吸附)精确控制的铂单原子层(PtASL)结构。
研究发现Pt1的局部配位环境不仅决定了其催化活性,还决定了其在还原活化过程中的结构演变。在低温催化一氧化碳(CO)氧化反应中,嵌入CeO2晶格中的PtASL活性位点表现出最高的转化频率,其活性是吸附在CeO2表面的PtASL位点的3.5倍,是Pt1位点的10-70倍。
进一步研究表明,嵌入的PtASL有利于CO的吸附和促进CeO2中晶格氧的活化/反应性,因而有效促进了CO催化氧化活性。这项工作为精确控制活性金属位点的局部配位结构,以及实现100%的原子利用效率和目标反应的最高本征催化活性提供了新思路。
背景介绍
由于具有近100%的金属利用效率,原子级分散的金属催化剂在多个应用领域中表现出巨大优势,尤其是对于高成本的贵金属催化剂。作为多相催化中的一个新兴材料体系,负载型金属单原子催化剂(SACs)在过去十年中得到了广泛的应用。
SACs的本征催化活性高度取决于孤立金属原子的配位结构,然而精确控制SACs中金属原子的配位结构仍然是一个巨大的挑战。此外,由于完全孤立的金属原子之间缺乏协同作用,提高SACs对特定反应比如低温CO氧化的本征活性则极为困难。
最近有研究表明,在有缺陷的载体上可以构筑金属原子单层(ASL)或二维筏(2D rafts)结构,该类结构不仅可以保持100%的金属利用效率,同时相邻金属原子之间可表现出协同效应,因此在多个催化应用中表现出独特的优势。除了在特定载体上直接合成二维筏结构之外,金属ASL结构也可以有效地通过对金属单原子的后还原处理来进行构筑。
例如,通过在H2气氛中的精确还原控制,Pt集合体(ensemble)或Pt单层结构可以由氧化铈(CeO2)上负载的Pt单原子制备获得,它们在CO氧化中表现出比Pt单原子催化剂更高的活性。尽管如Pt等金属ASL结构已经在多个载体上成功地制备,但据我们所知,这些结构通常存在于载体表面,相对缺乏与载体晶格之间的强相互作用。
据报道,无论对于负载型金属单原子还是金属团簇催化剂,金属活性位的局部配位结构决定了其催化性能。然而,对于金属ASL催化剂,ASL局部配位结构是否同样影响其催化性能仍不清楚。此外,以Pt为例,如果Pt ASL活性位可以由Pt SACs经过后还原处理制备得到,那么Pt SACs的配位结构是否会影响最终形成的Pt ASL活性位的局部结构、进而影响其催化活性仍需要进行系统的研究。
研究目标
原子尺度金属位点局部配位结构的精确调控以及CO氧化反应中活性位催化行为的揭示。
图文精读
通过初湿浸渍法制备了负载量为30 wt% CeO2的CeO2/Al2O3 (CA)载体,并通过H2还原处理来进行缺陷富集。使用最佳条件制备的缺陷富集的CA载体可命名为CA-HD(HD表示“高密度”缺陷)。如Figure 1A-1B中的AC-STEM电镜照片所示,在CA和CA-HD载体上成功制备了具有0.25 wt% Pt的Pt单原子(Pt1)催化剂,这些Pt1催化剂分别命名为Pt1/CA和Pt1/CA-e(-e表示CA-HD上“嵌入”的Pt物种)。
In situ DRIFTS of CO adsorption结果发现CO线性吸附于Pt1/CA和Pt1/CA-e上的Pt1位点(Figure 1C),且表现出不同的吸附波数(2096 cm-1和2092 cm-1)。XANES结果(Figure 1D)表明Pt1/CA和Pt1/CA-e中的Pt物种处于更接近于PtO2中的氧化状态,EXAFS结果证实Pt1/CA和Pt1/CA-e中不存在Pt-Pt金属键或Pt-O-Pt键,只有归属于Pt1结构的Pt-O和Pt-O-Ce(Figure 1E)。
这些结果清楚地表明,Pt物种在Pt1/CA和Pt1/CA-e催化剂中均以单原子形式存在,但具有不同的局部配位环境。
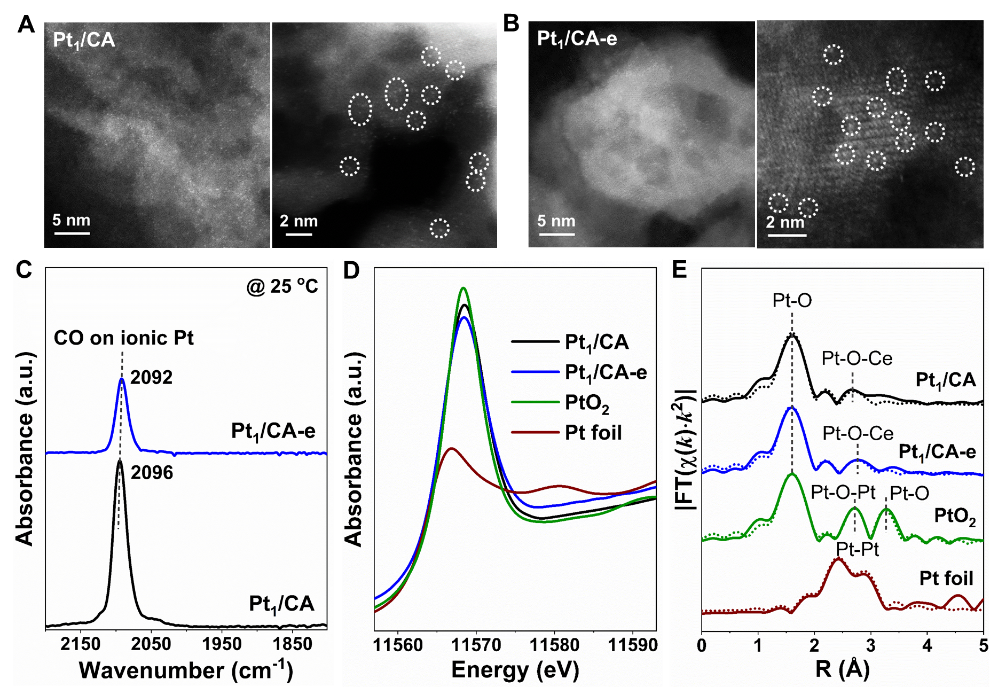
Figure 1. Structural characterization of Pt SACs. (A, B) High-angle annular dark field (HAADF) aberration-correction scanning transmission electron microscopy (AC-STEM) images and (C) in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of CO adsorption on Pt1/CA and Pt1/CA-e catalysts; (D) normalized Pt L3-edge X-ray absorption near edge structure (XANES) and (E) Fourier-transformed k2 - weighted extended X-ray absorption fine structure (EXAFS) oscillations in R space for the Pt L3-edge in Pt1/CA and Pt1/CA-e catalysts (solid line: experimental data; dotted line: fitted data).
Figure 2A-2B显示了Pt1/CA和Pt1/CA-e催化剂上CO吸附随时间变化的情况。当室温下暴露于CO气氛中时,在Pt1/CA上观察到2096和2120 cm-1波段的吸附峰逐渐增强,说明CO可同时以化学吸附和物理吸附的状态持续吸附于Pt1位点。不同的是,在Pt1/CA-e上观察到,只有当Pt1位点上物理吸附的CO (2120 cm-1) 达到饱和状态后,2092 cm-1处的化学吸附峰才逐渐出现,表明嵌入的Pt1位点上CO的化学吸附可能只有当Pt1位点被物理吸附的CO改变后才会发生。为了进一步证实这一猜测,我们对这一过程进行了DFT模拟计算。
如Figure 2C所示,当Pt1附近没有额外的氧空位时,在嵌入的Pt1(O)4结构上只能实现CO物理吸附(Eads = -0.34 eV);一旦嵌入的Pt1(O)4结构中的氧被物理吸附的CO还原去除(ΔE = 0 eV,Ea = 1.15 eV),较强的CO化学吸附(Eads = -1.99 eV)则会在该Pt1上发生。
由于在CO化学吸附后两个Pt1-O键很容易发生断裂,因此该状态下Pt1与载体键合较弱并容易发生扩散(形成Figure 2C中的结构iv)。随着Pt1(CO)结构和附近表面氧原子的有利扩散,最终会形成稳定的带有表面氧空位(Ov)的Pt1(O)3(CO)结构(即Figure 2C中的结构vi)。
在该结构上计算得到的CO吸附波数为2091 cm-1,与实验测量(2092 cm-1)和文献报道的结果一致。因此,通过多种表征方法和DFT计算证实,Pt1/CA和Pt1/CA-e催化剂上的Pt1物种的确具有不同的局部配位环境,它们分别是CeO2 (100)表面上吸附的Pt1(O)4结构以及表面缺陷富集的CeO2 (100)表面上嵌入的Pt1(O)4结构。
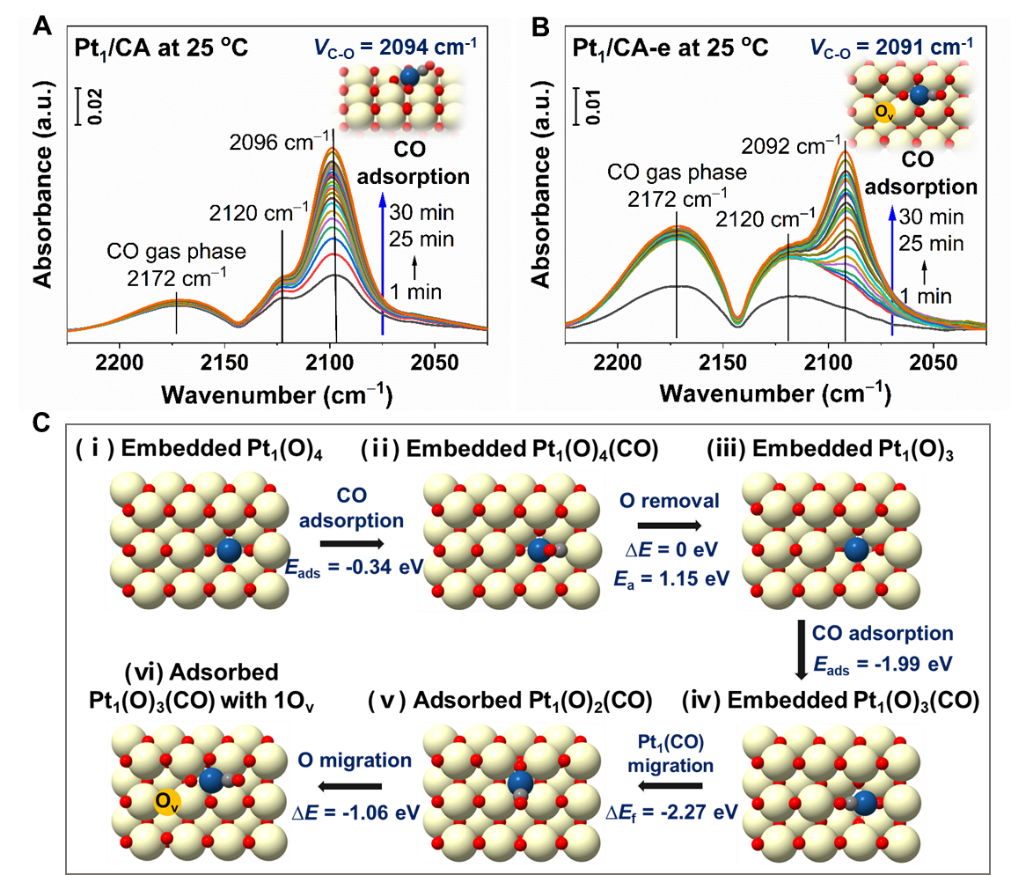
Figure 2. Time-resolved CO adsorption and theoretical calculations of Pt1 structures after CO adsorption. In situ DRIFTS of CO adsorption as a function of exposure time in CO flow on (A) Pt1/CA and (B) Pt1/CA-e; as well as (C) simulated structure evolution process of Pt1 within Pt1/CA-e catalyst during CO adsorption. Color code: Ce (yellow), O (red), Pt (blue).
通过精细调控H2还原条件,以Pt1/CA和Pt1/CA-e作为前驱体在CA和CA-HD载体上成功构筑了对CO氧化最具活性的Pt位点。通过AC-STEM技术对两个催化剂表征发现,最活性的Pt位点是在CeO2 (020)上负载的具有100% Pt原子分散的Pt原子单层结构(PtASL)(Figure 3A-U)。
从Pt1/CA活化获得的催化剂上(命名为PtASL/CA)观察到,具有Pt-Pt间距(dPt-Pt)大约为2.8 Å的PtASL结构吸附于CeO2表面(Figure 3I,J),未发现PtASL和CeO2之间存在明显的晶格匹配关系。对于由Pt1/CA-e活化获得的催化剂(命名为PtASL/CA-e),不同于PtASL/CA催化剂,发现dPt-Pt为2.6 Å的PtASL结构以取代Ce原子的形式嵌入于CeO2表面晶格(Figure 3T,U),并与CeO2之间具有完美的晶面匹配关系。
如Figure 3V,W所示,在PtASL结构上清楚地观察到线性(2078和2076 cm-1)和桥式(1841和 1840 cm-1)吸附的CO物种。以上结果表明,Pt1在CA和CA-HD上不同的局部配位结构决定了还原活化过程中不同的Pt1演化过程。
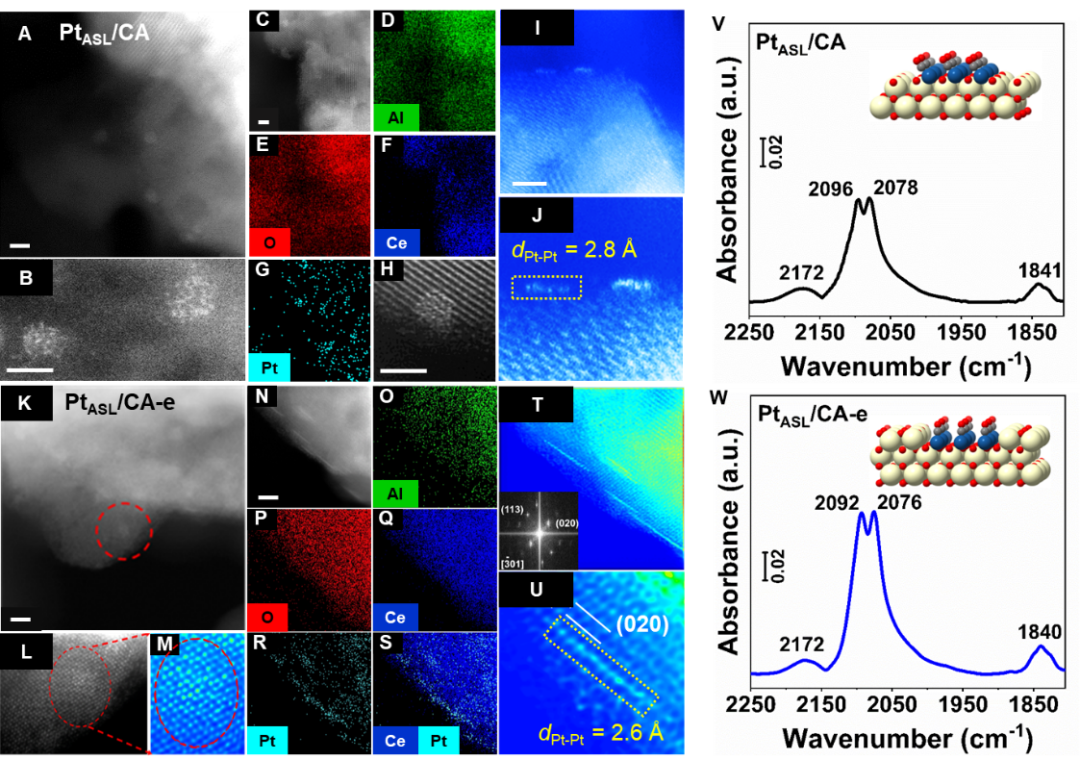
Figure 3. Structural characterization of Pt atomic single-layer catalysts. HAADF AC-STEM images and EDS mapping results (with scale bars as 2 nm) for (A-J) PtASL/CA and (K-U) PtASL/CA-e, and in situ DRIFTS of CO adsorption on (V) PtASL/CA and (W) PtASL/CA-e in CO flow at 25 oC (the models of PtASL/CA and PtASL/CA-e with adsorbed CO are shown as inserted in V and W).
Figure 4显示了具有不同Pt结构(吸附或嵌入型Pt1和PtASL)的Pt1/CA、Pt1/CA-e、PtASL/CA和PtASL/CA-e催化剂的CO氧化活性。如Figure 4A 所示,Pt1/CA表现出比Pt1/CA-e更高的催化CO氧化活性。活化后,具有PtASL结构的PtASL/CA和PtASL/CA-e催化剂比其相应的Pt1前驱体催化剂表现出更加优异的CO氧化活性。
其中,PtASL/CA-e催化剂的CO氧化活性明显优于PtASL/CA催化剂,前者达到50% CO转化率时的温度远低于后者。在这些催化剂中,具有嵌入型PtASL结构的PtASL/CA-e催化剂表现出最好的低温CO氧化活性(Figure 4A)。此外,发现H2O的加入可明显促进PtASL/CA和PtASL/CA-e催化剂对CO催化氧化的活性(Figure 4B),在加入5% H2O的情况下,PtASL/CA-e催化剂仍然表现出比PtASL/CA高得多的CO氧化活性。
动力学研究发现,这些催化剂上CO氧化的表观活化能(Eapp.)按Pt1/CA-e(77 ± 3 kJ/mol)>Pt1/CA(68 ± 3 kJ/mol)>PtASL/CA(50 ±2 kJ/mol)>PtASL/CA-e(43 ± 2 kJ/mol)的顺序依次降低(Figure 4C),再次证实了CeO2上嵌入的PtASL结构是所有Pt结构中对CO氧化最为活泼的位点。
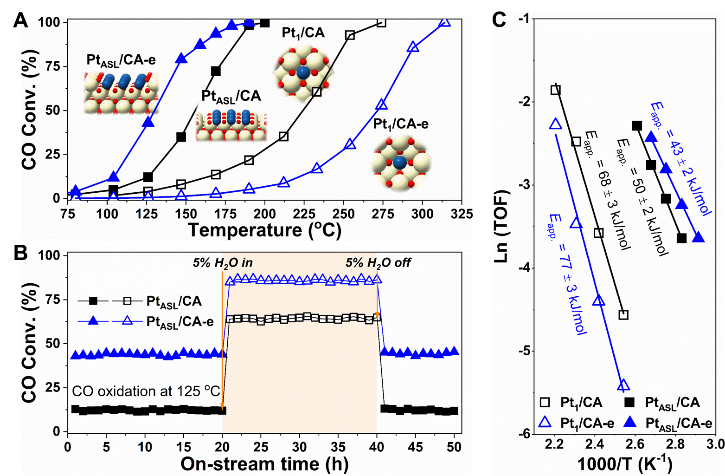
Figure 4. Catalytic performance measurements. CO oxidation activity on (A) Pt/CA catalysts with different Pt structures and (B) PtASL/CA and PtASL/CA-e catalysts with H2O in feedstock, and (C) Arrhenius plots and apparent activation energy (Eapp.) values for CO oxidation on Pt/CA catalysts with different Pt structures. Reaction condition: Steady-state testing, [CO] = [O2] = 1%, [H2O] = 5% (if used), balanced with Ar, with WHSV of 200,000 mL·gcat-1·h-1.
为了确定催化剂中的活性氧物种,利用in situ DRIFTS耦合MS技术实时监测了CO与PtASL/CA和PtASL/CA-e催化剂上的氧物种在100 °C时的反应状况。如Figure 5A所示,与PtASL/CA上观察到的前2分钟内较强的CO2生成峰相比,由于存在较少的表面氧物种,在PtASL/CA-e上观察到了较低的CO2生成峰。
然而,一旦吸附型PtASL结构中的表面氧物种被大量消耗,在PtASL/CA上CO2的生成就会急剧下降;不同的是,PtASL/CA-e开始表现出较低的CO2生成量,但随着CO吸附时间的延长(2分钟以上),它上面CO2的生成量则明显高于PtASL/CA。
这些结果表明,与嵌入型PtASL结构相关的晶格氧比与吸附型PtASL结构相关的晶格氧更具反应活性。独特的嵌入型PtASL结构可以更有效地促进CeO2中晶格氧的活化,这使得PtASL/CA-e催化剂表现更为优异的CO氧化活性。如Figure 5B所示,PtASL/CA和PtASL/CA-e催化剂上CO吸附物种随着时间逐渐累积,而Figure 5C则显示了在100°C时PtASL/CA和PtASL/CA-e催化剂上不同Pt位点上CO吸附强度与CO2生成之间的关系。
在两种催化剂上都观察到,随着CO2生成的减少PtASL位点上吸附的CO(2069 cm-1)逐渐增加,然而Pt1位点上的CO吸附(2086 cm-1)没有明显变化。这些结果表明,吸附在PtASL位点上的CO分子相对于吸附在Pt1位点上的CO分子更具反应活性。
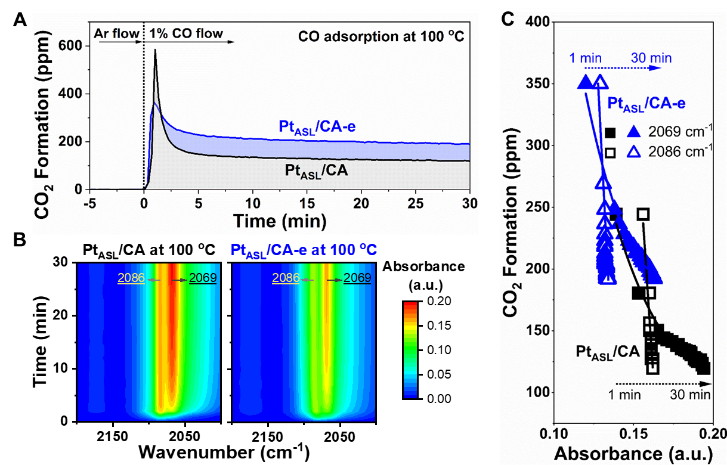
Figure 5. Measurements on active oxygen species. Time-resolved (A) CO2 formation after exposure to continuous CO flow and (B) in situ DRIFTS of CO adsorption (in CO flow only), and (C) CO2 formation as a function of CO adsorption intensity resulting from the reaction between CO and oxygen species within PtASL/CA and PtASL/CA-e catalysts
为了进一步解析PtASL/CA-e催化剂具有优异CO氧化性能的原因,利用DFT计算对嵌入型PtASL结构上的CO氧化反应机理进行了研究。如Figure 6所示,以被*CO完全覆盖、带有缺陷的嵌入型PtASL结构(具有两个表面Ov)作为起始结构(结构i),提出了详细的CO氧化机制。
首先,气相O2分子以-0.47 eV的吸附能占据表面氧空位(Ov)(结构ii)。吸附的*O2随后会激活界面上的O,使其Ov形成能从2.25 eV大幅降低到0.48 eV。相比之下,同一结构(结构ii)上Pt-O-Pt和Pt-O-Ce中O的Ov形成能分别为1.44和2.09 eV,因此推测参与CO氧化的O物种主要来自CeO2界面。
进一步计算发现,PtASL上吸附的*CO与该界面O反应的活化能为0.62 eV(TS-1)。在释放第一个CO2分子(结构iii)后,CO吸附的Pt位点会伴随着界面氧空位的形成而重新暴露。随后,气相CO分子以1.57 eV的放热吸附于该Pt位点(结构iv)。
此后,化学吸附的*O2被CeO2界面上的Ov激活,使其具有1.45 Å的O-O键长(结构iv),然后解离形成一个表面*O和一个界面*O(结构v)。该O2解离步骤的活化能和反应能分别计算为0.71和0.25 eV。一旦界面*O恢复,*CO将和界面*O发生第二次CO氧化,其能垒计算为0.57 eV(TS-3)。
由于前一个*O2解离步骤有0.25 eV的吸热,修正后的第二次CO氧化表观活化能为0.82 eV。在CO2脱附后(结构vi),气相CO可再次吸附到暴露的Pt原子上(结构 vii)。最后,表面*O以0.44 eV的放热迁移到界面Ov处(~0 eV能垒),实现催化剂再生。
在整个过程中可以发现,PtASL/CA-e上催化CO氧化的决速步骤为O2分子的解离和第二次CO氧化(从结构iv到vi),其表观活化能为0.82 eV。进一步详细计算发现,PtASL/CA-e上的CO氧化活化能远低于PtASL/CA上的活化能(0.82 eV比1.78 eV),因为PtASL/CA-e催化剂比PtASL/CA催化剂在低温CO氧化反应中表现出了更为优异的催化活性。
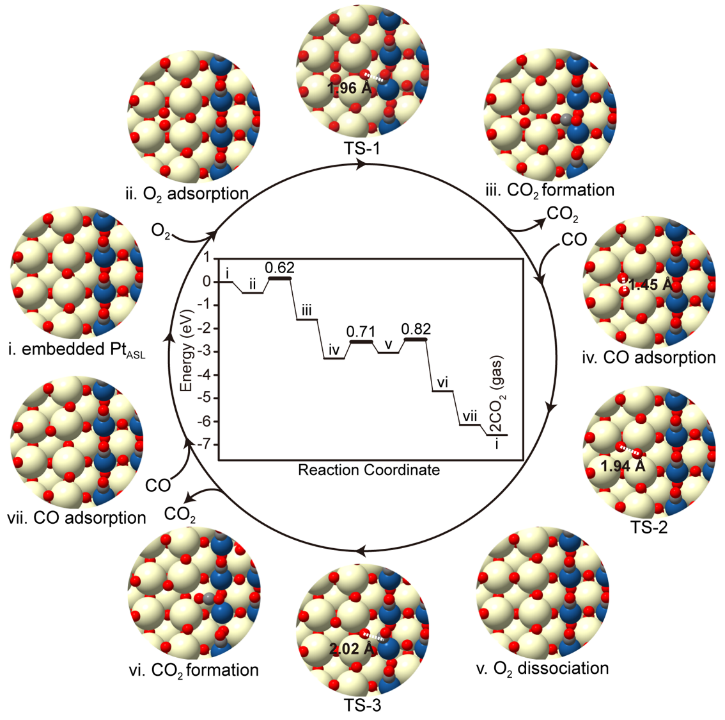
Figure 6. Potential energy diagrams and configurations for the CO oxidation cycle on PtASL/CA-e catalyst. The CO oxidation proceeded on an embedded Pt atomic single-layer unit in PtASL/CA-e. The reaction energies and activation energies are indicated in eV in the potential energy diagram. Color code: Ce (yellow), O (red), Pt (blue), C (gray).
心得与展望
通过控制初始Pt单原子的配位环境和活化条件,本研究实现了具有不同配位环境的Pt单原子层的可控制备。基于可控制备的Pt结构,揭示了Pt/CeO2基催化剂上对CO催化氧化反应最优的Pt活性位点。下一步研究可扩展到配位环境可控的Pt团簇催化剂或其他金属单原子层/团簇催化剂的合成、深入表征以及在其他催化反应中的应用。
审核编辑:刘清
- 相关推荐
- 热点推荐
- 催化剂
-
可控源音频大地电磁测量法在砂岩型铀成矿地质环境研究中的应用2011-03-04 1968
-
碱性醇类燃料电池新型催化剂的研究2011-03-11 1914
-
白光LED结构化涂层制备及其应用研究2022-03-29 10894
-
单分散纳米微粒制备方法研究进展2010-01-02 782
-
双向可控硅液位控制器电路2008-11-07 5483
-
双向可控硅液位控制电路2009-07-27 1001
-
N+缓冲层对PT-IGBT通态压降影响的研究2013-05-06 1088
-
石墨烯量子点的介绍、制备、表征与应用研究2016-12-08 6020
-
高质量二维原子晶石墨烯的制备及性能研究2017-10-27 839
-
高活性生物质碳负载Fe/Pt单原子双功能催化剂开发2021-02-12 3301
-
盐模板辅助制备多种过渡金属单原子催化剂用于高效氧还原2022-08-11 2125
-
石墨烯制备新技能:超临界流体技术2023-07-06 2317
-
什么是原子层刻蚀2025-01-20 1302
-
详解原子层沉积薄膜制备技术2025-05-14 1214
-
原子层沉积(ALD)制备高透光掺铌SnO₂电子传输层(ETL)实现高效钙钛矿太阳能电池2025-05-28 1043
全部0条评论

快来发表一下你的评论吧 !
