

非晶态氟硫酸铁电极具有稳定可逆性的插层和转化反应
描述
一、全文概要
锂离子电池(LIBs)已经成为电动汽车和可再生能源大规模储能系统被广泛接受和使用。然而,决定这些应用持续成功的渐进式改进依赖于先进的LIB技术的发展,即具有更高的能量密度和更低的成本的双重属性。目前LIB技术在能量密度和成本方面的限制主要与基于插层的过渡金属氧化物的正极化学有关,如LiNi1-x-yCoxMnyO2(0≤x,y≤1),其实际容量正在迅速逼近其理论界限。为了实现未来电池理想的大容量性能和低成本生产的双重特性,在电极中利用丰富储量的过渡金属进行的多氧化还原反应具有很重要的意义。
二、正文部分
1、成果简介
韩国首尔大学Kisuk Kang及其团队制备了一种非晶态的氟硫酸铁电极:a-LiFeSO4F,该材料可以利用具有稳定可逆性的插层和转化反应进行高效电化学反应。a-LiFeSO4F电极的容量为360 mAh g-1,即使在高温(60℃)下200次循环后仍有约98.6%的容量保持率。与传统的插层/转化型电极相比,本研究的新电极的高可逆循环稳定性归功于a-LiFeSO4F固有的非晶态结构,其结构完整性即使在转化反应后也不会受到严重干扰,从而使其能够继续作为插层宿主继续存在。本研究认为,插层/转化反应的这种循环稳定性可以普遍推广到各种非晶态插层材料,为利用多机制的锂化过程为设计高容量电极提供新的见解。
2、研究亮点
1)制备了一种非晶态的氟硫酸铁电极,:a-LiFeSO4F。该材料可以利用具有稳定可逆性的插层和转化反应进行高效电化学反应。
2) LiFeSO4F电极的容量为360 mAh g-1,即使在高温(60℃)下200次循环后仍有约98.6%的容量保持率。
3、图文导读
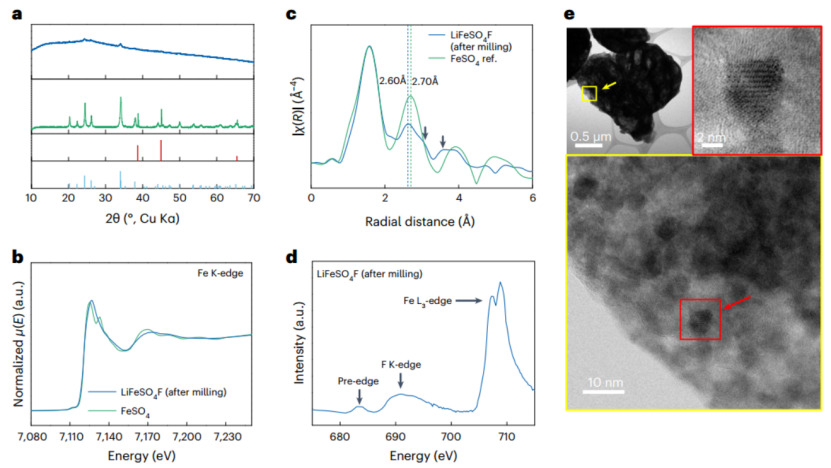
图1|a-LiFeSO4F的表征。a,LiF-FeSO4研磨后(蓝色)和研磨前(绿色)的XRD图,以及LiF(红色)和FeSO4(浅蓝色)的参考Bragg峰。 b,a-LiFeSO4F(蓝色)和FeSO4(绿色)的XAS Fe K-edge光谱。c,a-LiFeSO4F(蓝色)和FeSO4(绿色)的围绕Fe的径向分布。d,准备好的a-LiFeSO4F的F K-edge和Fe L3-edge。e, a-LiFeSO4F的扫描透射电子显微镜(STEM)亮场(BF)图像。
a-LiFeSO4F材料是由含有化学计量的LiF和FeSO4(摩尔比=1/1)的前体混合物与另外20wt%的石墨导电剂在氩气环境下通过简单的机械化学反应合成的。图1a显示了球磨过程前后样品的X射线衍射(XRD)图案,表明含有结晶LiF和FeSO4前体的混合物在球磨后发生了明显的非晶化。图1b中的Fe K-edge光谱表明,a-LiFeSO4F和FeSO4的近边X射线吸收精细结构是可以被明显区分的,特别是在大约7127-7200 eV处,这表明a-LiFeSO4F中Fe附近的局部结构发生重新排列。然而,两相的白线在同一位置出现,证实了a-LiFeSO4F产物中的Fe2+氧化状态。图1c中围绕铁的径向分布分析也阐明了a-LiFeSO4F的独特局部结构,它与原始的FeSO4相不同。如图1c中的箭头所示,邻接键的变化是可以清楚地检测到的,在大约3.01Å和3.60Å处有额外的肩峰。此外,a-LiFeSO4F的第二邻接键距离(~2.6Å)比原始FeSO4相(~2.7Å)略短。在图1d的SXAS中,新的氟键合峰的演变也可以从氟的K-edge检测到产品相(a-LiFeSO4F)。该光谱证明了在球磨过程后出现的683.5 eV的预边峰的存在。此外,图1d中几乎检测不到SXAS光谱中约700 eV的LiF信号的指纹,证明了在a-LiFeSO4F的形成过程中LiF前体的消耗。图1e中的透射电子显微镜(TEM)图像显示,该产物由微米级的颗粒组成,这些颗粒由随机分布的4-8纳米大小的颗粒组成。
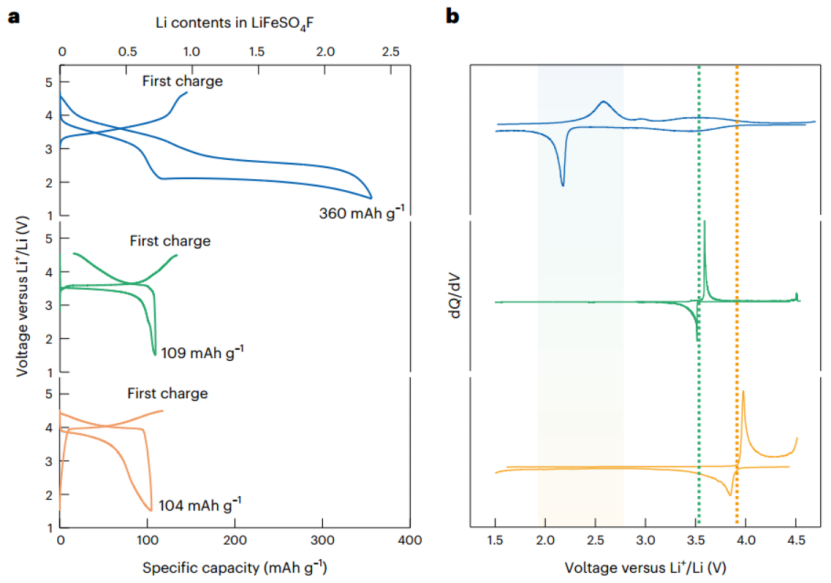
图2 |a-LiFeSO4F和多晶型物质(tavorite and triplite)的氧化还原活性。(a)在60℃、40 mA g-1条件下的充放电曲线以及作为电压函数的容量差(dQ/dV曲线)。(b)在60℃、40 mA g-1条件下的a-LiFeSO4F(蓝色)、tavorite(绿色)和triplite(橙色)的充放电曲线。
图2a显示了a-LiFeSO4F电极在第一次充电和随后的放电/充电过程中的电化学曲线)。为了进行比较,本研究还构建了由tavorite-LiFeSO4F和triplite-LiFeSO4F电极组成的电化学电池,其电化学曲线分别显示在图2a的中间和底部。与tavorite和triplite的情况相比,该区域的电压曲线似乎大大倾斜,这归因于进入非晶态结构的特征锂化现象。这种性质在图2b的差分(dQ/dV)曲线中显示得更加清楚。尽管在~3.60V和~3.90V的电压下,观察到tavorite和triplite-LiFeSO4F电极有尖锐的氧化/还原峰,但a-LiFeSO4F电极在类似的电压范围内没有表现出明显的峰值。
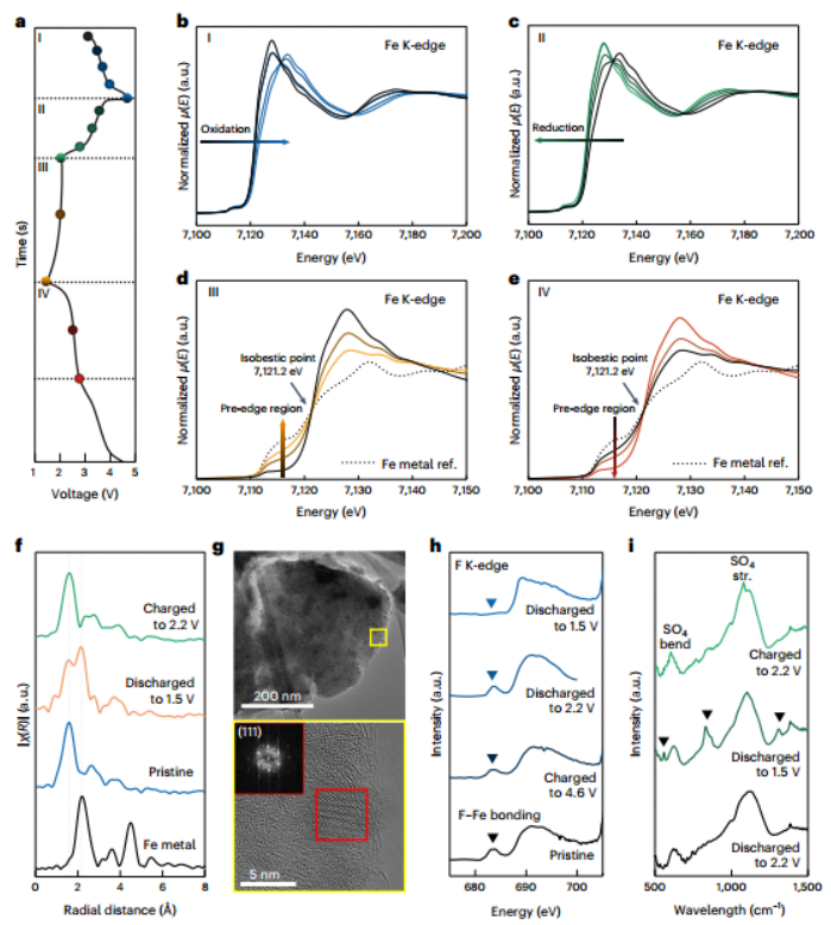
图3 |用于机理研究的异位分析。a,a-LiFeSO4F的电压与时间的充电/放电曲线。b-e,高压区充电(b)和放电(c)以及低压区充电(d)和放电(e)的Fe K-edge X射线吸收近边缘结构光谱。f,参考Fe金属、原始状态、放电到1.5V和充电到2.2V的Fe K-edge EXAFS光谱。g,完全放电状态的TEM图像和快速傅里叶变换(FFT)和衍射图案。i,半放电、完全放电和半充电状态的红外(IR)光谱。在~603 cm-1处的峰对应于SO4的弯曲振动,而在~1107 cm-1处的峰对应于SO4的拉伸(STR)振动。
本研究通过探测局部结构演变和铁的价态变化来研究充电/放电过程,如图3所示,尽管在整个过程中由于其非晶态的性质,晶体结构的变化不能被清楚地检测到。图3a-e显示了电极在高压区(2.2-4.7V,图3a I,II)和低压区(1.5-2.2V,图3a III,IV)的第一次充电和随后的放电/充电循环期间的原位硬X射线吸收光谱。如图3b,c所示,在高电压区域,Fe K-edge白线随着充电和放电分别转移到更高和更低的能量值,表明该区域的电化学活性源于典型的脱层/插层过程中的Fe2+/3+氧化还原反应。本研究观察到:在进一步放电到低电压区域时,a-LiFeSO4F的XAS光谱在轮廓形状上发生了明显的变化,并表现出Fe0金属相的特性,如图3d所示。此外,在随后的充电过程中也可以观察到它的可逆性,如图3e所示。图3f中的EXAFS分析进一步支持a-LiFeSO4F电极的可逆转换反应。2.2 Å处的Fe-Fe键对应于原始a-LiFeSO4F电极放电(~1.5 V)时出现的Fe金属相,但在重新充电(~2.2 V)时会减弱。图3g中的TEM结果直接验证了放电状态下嵌入锂化合物基体中的纳米级(5-8 nm)铁的存在。本研究还关注了氟的局部结合的变化,以进一步阐明发生在低电压区域的转换反应,如图3h所示。图3h说明原始电极中F-Fe键合的前缘(三角形符号)在高电压区域(》2.2V)保持不变,但在放电到1.5V时明显有减弱,这与转换反应中F-Fe键合的解离一致。
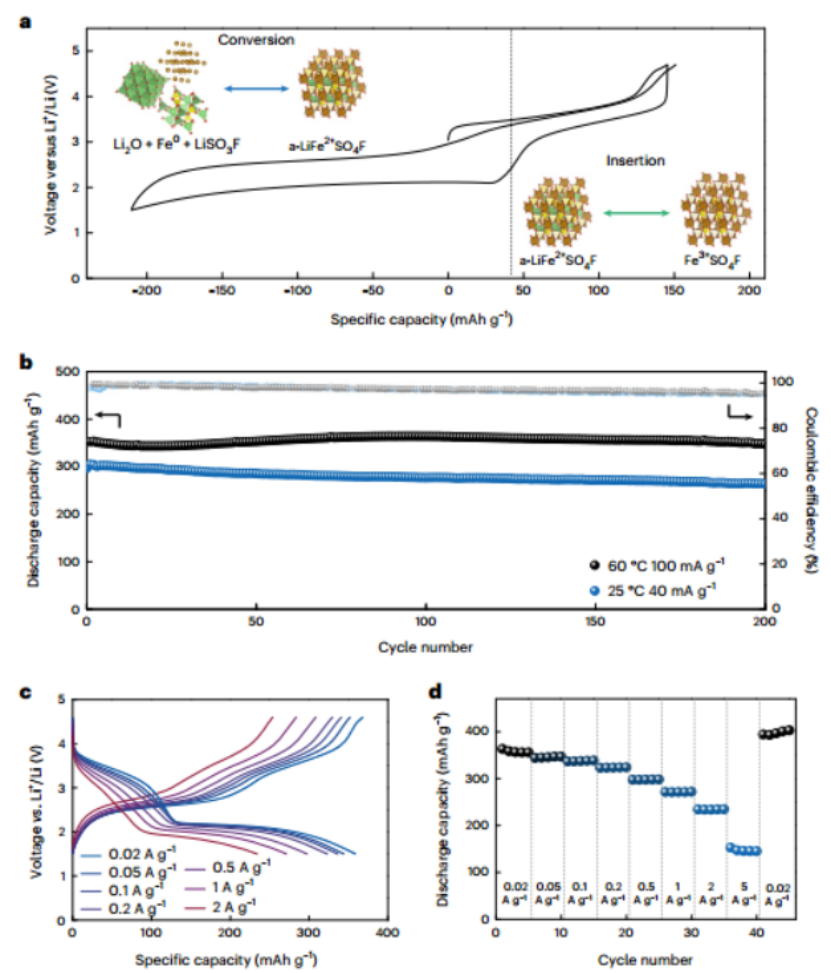
图 4 |a-LiFeSO4F的电化学性能。a,a-LiFeSO4F的充电/放电曲线以及插入和转换反应的两步反应机制示意图。b,在25℃和60℃,电流密度为40 mA g-1时测量的容量保持率和库伦效率与循环数的关系。c,电流密度为0.02 A g-1、0.05 A g-1、0.1 A g-1、0.2 A g-1、0.5 A g-1、1 A g-1和2 A g-1时a-LiFeSO4F的充/放电曲线。
图4a显示了a-LiFeSO4F电极的电化学反应示意图以及特征性的充电/放电曲线,它显示了基于联合插层和转换的三电子氧化还原反应。图4b显示了a-LiFeSO4F电极在扩展的充电和放电循环中的电化学稳定性。电极表现出超过300 mAh g-1的容量,在室温下进行的实验表明:其能够稳定地进行200个循环,容量保留率为90%。图4c,d显示了a-LiFeSO4F电极在不同电流率下的功率输出能力。当电流密度从20增加到5000 mA g-1时,可提供的比容量系统地减少。然而,即使在1000 mA g-1的电流密度下,仍然可以保留超过250 mAh g-1的比容量,这证明其具有不错的倍率性能。由于反复的转化反应会引起形态上的变化,导致电极结构的轻微重组,在20 mA g-1的最后循环中,容量的增加将归因于电极中最初不活跃的a-LiFeSO4F颗粒的激活。
4、总结与展望
非晶态的LiFeSO4F被认为是一种高容量的电极材料,可以成功地利用插层反应和转换反应。这种材料可以提供360 mAh g-1的可逆容量,对应于Fe2+/3+插层反应的一个电子(110 mAh g-1)转移和Fe0/2+转换反应的一个双氧化还原(250 mAh g-1)。它具有200次循环的出色的循环稳定性,在25℃和60℃下容量保持率分别为90%和98.6%。观察到的可逆反应被归因于a-LiFeSO4F的非晶态特性,因为固有的缺陷的非晶态结构对通常伴随着重复转换/再转换反应的潜在结构紊乱而言是免疫的。此外,原始a-LiFeSO4F相和放电产物的非晶态性质提供了富含空位的环境,在这种环境中,金属(Fe2+)跨越体/界面的迁移比结晶相中的迁移更容易,从而促进了还原/转换电极动力学的发展。包括插层和转换反应在内的高度可逆的多机制锂化过程的成功展示意味着在各种非晶态插层材料中可以更普遍地实现高容量特性。在这个方向上的进一步研究将提供新的机会来解决以低成本获得高容量的难题,使传统的基于插层的电极的容量上限被打破。
审核编辑:郭婷
-
不同浓度和温度的硫酸对材料的腐蚀差别较大2013-01-10 0
-
不可逆布雷顿制冷循环的性能优化2010-01-01 500
-
硫酸渣制备铁红工艺流程2009-03-30 1088
-
非晶态结构2009-08-06 2655
-
宁波国际非晶态材料的制备方法2020-04-19 2271
-
非晶态半导体阈值开关的机理2021-03-24 2399
-
硫酸亚锡在电池中的反应原理与具体作用是什么2021-05-24 4000
-
解析用于超稳定Zn电池的高产率碳点中间层2022-08-08 541
-
如何准确评估实际锂金属电池的可逆性呢2022-11-08 1016
-
打开稳定低温锂金属负极的极化和可逆性限制2023-04-04 1121
-
离子在非晶态材料内的投影射程2023-05-15 1438
全部0条评论

快来发表一下你的评论吧 !
