

使用基于Mo矿物水凝胶设计不含碳的单个铁原子分散的异质结构纳米片
描述
研究背景
氢气因具有能量密度高、在空气中燃烧时排放的污染物少等优点而成为一种有备受关注的能源。在各种制氢方法中,电化学方法是较容易和更经济的。其中商业贵金属基HER电催化剂的高成本促使研究人员开发具有高活性和高稳定性的低成本电催化剂。具有单金属原子分散体(SACs)的异质结构材料是制氢的理想催化剂材料。然而,大规模制备高稳定性和低成本异质结构锚定单原子的催化剂仍然是一个巨大的挑战。
成果简介
鉴于此,香港城市大学吕坚院士、李扬扬,哈尔滨工业大学孙李刚(共同通讯作者)等提出使用基于Mo矿物水凝胶作为前体来设计新的不含碳(C)的单个铁原子分散的异质结构纳米片作为析氢催化剂。Fe/SAs@Mo基HNSs在HER中表现出优异的电催化活性和长期耐久性:在10mA cm-2下的过电位为38.5 mV,Tafel斜率为35.6mV dec-1,在200mA cm-2下可连续运行600 h。
研究亮点
1、通过低温磷化自组装无机-无机配位的FePMoG纳米片,得到了一种高效的HER电催化剂Fe/SAs@Mo基HNSs;
2、Fe/SAs@Mo基HNSs的异质结构界面和单一分散的铁原子有利于HER过程中H2O的吸附和适当的H*解吸附。
图文介绍
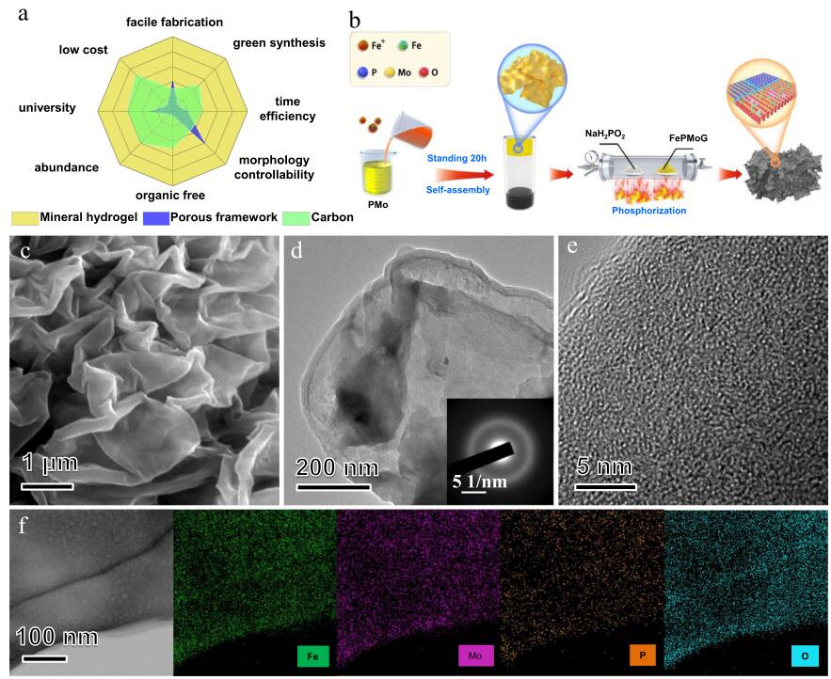
图1 制备FePMoG和Fe/SAs@Mo基HNSs电催化剂的概念设计和微观结构表征。
矿物水凝胶由无机物、无机盐和水制成,可以在温和条件下通过快速简单的混合和胶凝过程自组装成各种形态的凝胶网络。在加热处理后,单独分散在凝胶网络中的配位金属离子可以转化成金属氧化物/磷化物/硫化物的异质结构。与其他常见的单原子基底前驱体(多孔框架和碳)相比,矿物水凝胶在合成路线、环境友好、生产效率、可调节性、原材料丰富性、可持续性、和成本效益方面显示出巨大的优势(图1a)。图1b显示了制备FePMoGs纳米片的新型配位诱导自组装工艺过程。首先在室温下简单混合多金属氧酸(PMo)和三价铁离子(Fe3+)的溶液。通过配位诱导的自组装过程,形成了均匀分布的FePMoG纳米片的悬浮液。25∶1摩尔比的Fe3+: PMo的混合物可以形成宽度为几微米,厚度约为50纳米,且表面光滑起皱的纳米片(图1c)。图1d中的FePMoG的TEM图像证明了FePMoG复合材料的二维结构,且SAED图案(图1d中的插图)为宽衍射环,证实了其无定形性质。HR-TEM图像(图1e)显示了无定形FePMoG中原子的无序排列。FePMoG的STEM图像和相应的EDS揭示了Fe、Mo、P和O原子在其结构中的均匀分布,表明它由Fe3+和PMo之间的均匀相互作用组成(图1f)。
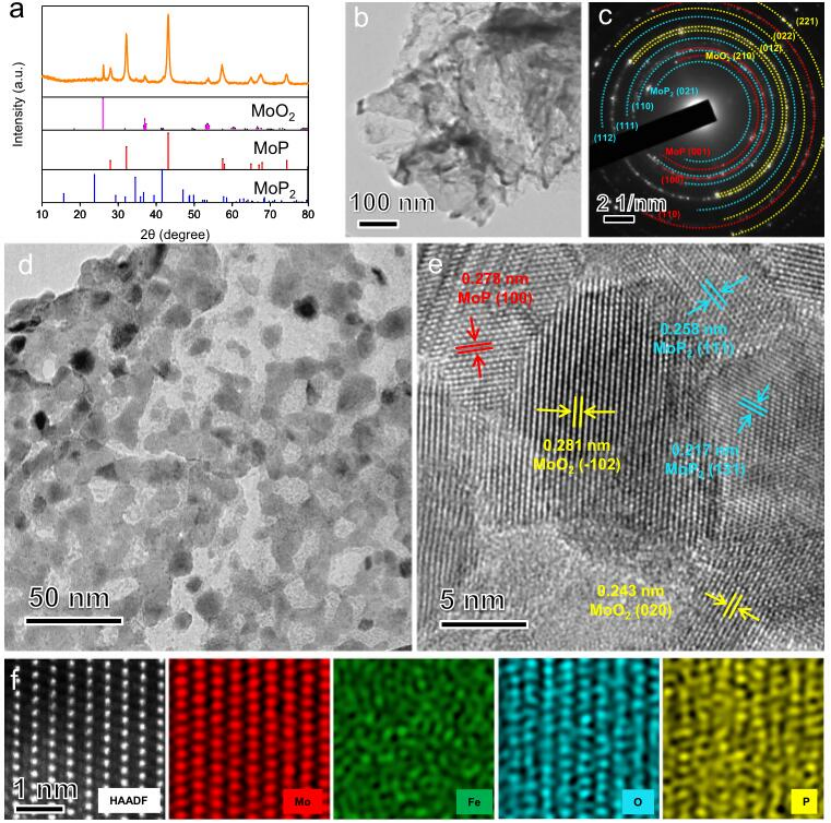
图2 Fe/SAs@Mo基HNSs的结构表征。
磷化后FePMoG纳米片的结构演变如图2所示。Fe/SAs@Mo基-HNSs的XRD图案表明存在二氧化钼(MoO2,JCPDS:78-1069)、磷化钼(MoP,JCPDS:240771)和二磷化钼(MoP2,JCPDS:89-2678)的特征衍射峰(图2a)。Fe/SAs@Mo基HNSs在磷化过程中保持其初始2D多孔纳米片形态(图2b)。相应的SAED图包含由MoO2,MoP,和 MoP2形成的环(图2c)。高倍率TEM图像显示Fe/SAs@Mo基-HNSs(图2d)包含平均直径<10 nm的孔的互连结构。图2e中的晶格条纹为0.2 81,0.243,0.278,0.209,0.258,和0.217 nm,分别对应于MoO2(102)、MoO2(020)、MoP(100)、MoP(101)、MoP2(111)和MoP2(131)的晶面间距,表明Fe/SAs@Mo基-HNSs的异质结构。Fe/SAs@Mo基-HNSs的HAADF-STEM图像和相应的EDS元素图揭示了Fe、Mo、P和O原子的均匀分布,并且Fe原子独立地分散在晶格中(图2f)。
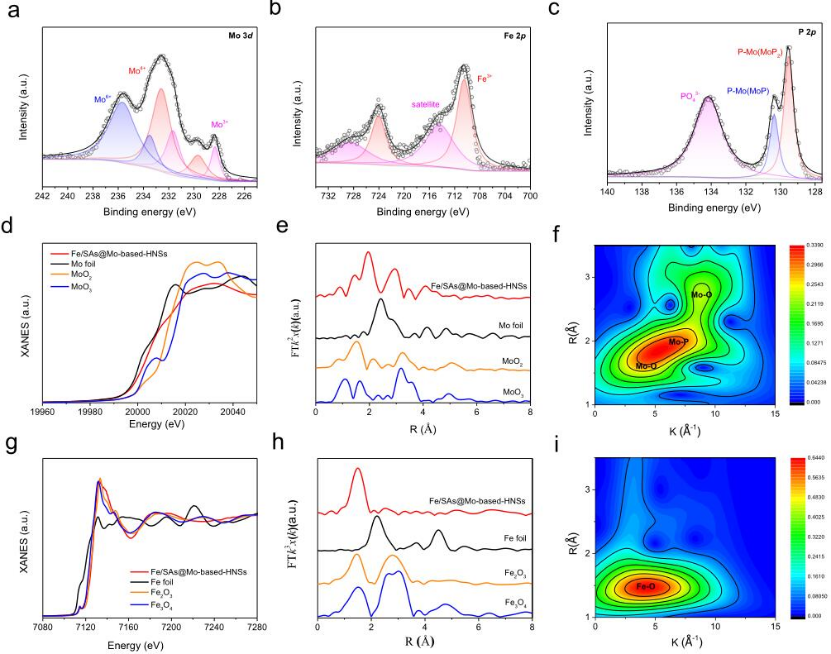
图3 Fe/SAs@Mo基HNS的XPS和XANES光谱。
采用XPS和XANES表征了Fe/SAs@Mo基-HNSs的化学组成、价态和配位状态以及电子结构(图3)。如图3a所示,Mo 3d的高分辨率XPS光谱由六个峰组成:231.66和228.34 eV (Mo3+)、232.58和229.68 eV(Mo4+)以及235.63和233.49 eV(Mo6+),分别对应于MoP、MoO2和MoP2。Mo K边缘XANES光谱显示Fe/SAs@Mo基HNSs的近边缘吸收能介于Mo箔和MoO2的吸收能之间,表明Mo的平均氧化态介于Mo0和Mo4+之间(图3d)。相应的EXAFS揭示了分散在Fe/SAs@Mo基HNSs中的Mo原子与P和O原子配位,不存在Mo-Mo键(图3e,f)。Fe的高分辨率XPS光谱(图3b)在710.7 eV (Fe 2p3/2) 和724.1 eV (Fe 2p1/2) 处具有Fe3+的特征峰,并且在715.6 eV处具有Fe3+2p卫星峰。Fe K边XANES光谱也显示Fe的化合价接近Fe3+(图3g)。相应的EXAFS结果描绘了铁原子的配位环境:在约1.5埃处的强峰归因于Fe-O键,在傅里叶变换EXAFS中,在2.47或2.85埃处不存在Fe–Fe峰(图3h),这表明Fe在Fe/SAs@Mo基HNSs中是孤立存在的,并且大部分与O进行配位(图3i)。
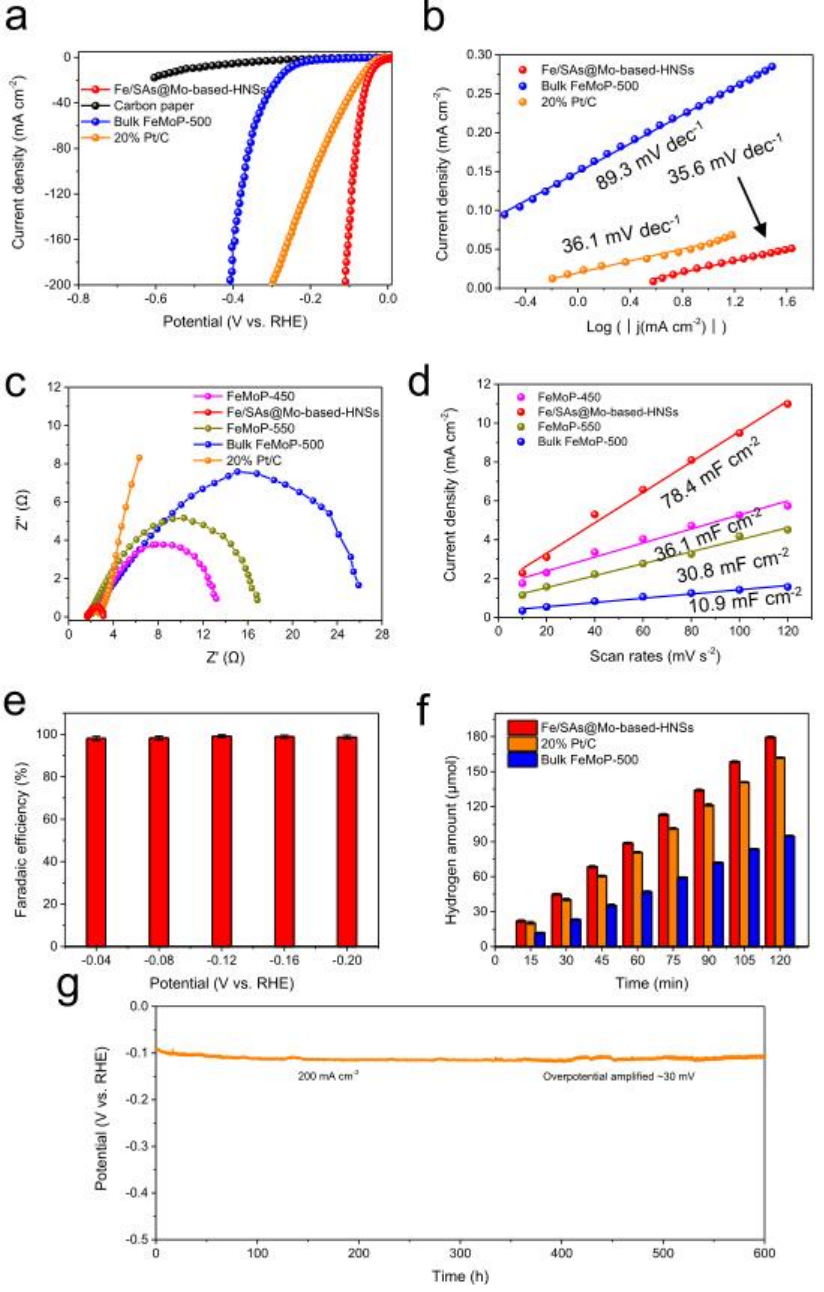
图4 Fe/SAs@Mo基HNSs在1.0 M KOH溶液中的HER电催化性能。
使用典型的三电极系统,在1 M KOH溶液中,研究了Fe/SAs@Mo基HNSs的HER性能。图4a所示的线性扫描伏安图表明Fe/SAs@Mo基-HNSs具有优异的催化性能:在10mA cm-2下,过电位仅38.5 mV,比FeMoP-T样品和商业20 wt% Pt/C的低得多。当电流密度达到200mA cm-2时,Fe/SAs@Mo基-HNSs的过电位略微增加到109.9 mV,显著低于商业20 wt% Pt/C的过电位(299.3 mV)。图4b表明Fe/SAs@Mo基HNSs的Tafel斜率(35.6mV dec-1)小于FeMoP-500(89.3mV dec-1)和其他FeMoP-T样品,而接近20% Pt/C(36.1mV dec-1)。如图4c所示,Fe/SAs@Mo基HNSs具有最小的电荷转移电阻。Fe/SAs@Mo基-HNSs的Cdl为78.4 mF cm-2(图4d),远高于直接磷化的块体FeMoP-500(10.9 mF cm-2)以及FeMoP-450和FeMoP-550样品的Cdl (分别为36.1和30.8 mF cm-2)。从图4e中可以看出,Fe/SAs@Mo基HNSs的法拉第效率在宽电势下接近100%。Fe/SAs@Mo基HNSs的H2产率随时间线性增加,并且显著高于商业20 wt% Pt/C和块状FeMoP-500的产率(图4f)。Fe/SAs@Mo基HNSs的稳定性也非常好:200mA cm-2的电流密度下可保持600 h(图4g)。
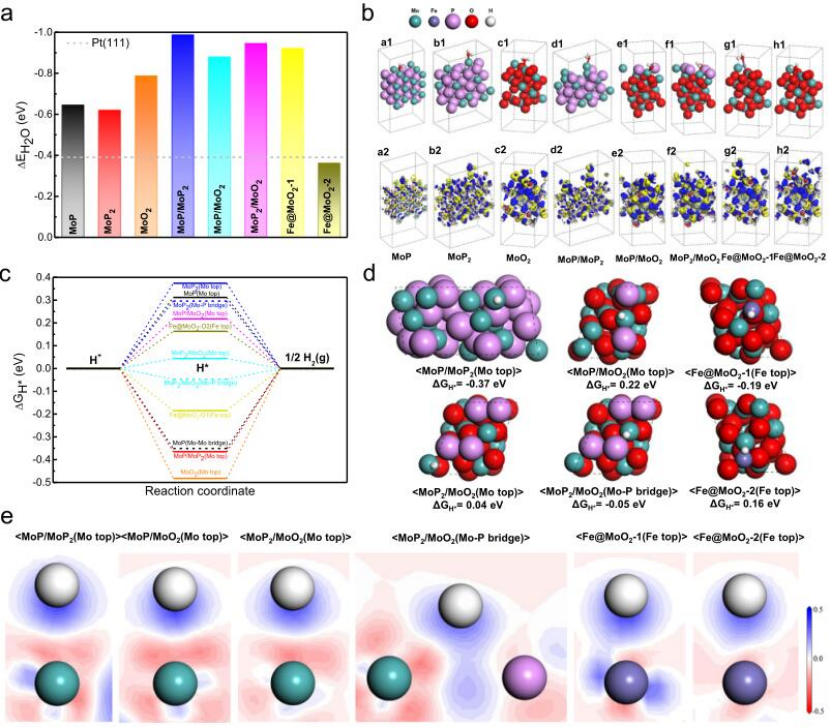
图5 设计用于在碱性条件下催化HER的Fe/SAs@Mo基HNS的DFT模拟。
采用DFT计算确定了Fe/SAs@Mo基HNSs在HER中的突出电催化活性的分子基础。计算了水分子在催化位点上的吸附能(ΔEH2O),图5a表明,所有界面模型都比单相模型具有更高的ΔEH2O值,证明异质结构界面有效地促进了H2O吸附。为了进一步解释增强H2O分子吸附的原理,DFT模拟的原子构型和在Mo或Fe位点的H2O吸附后相应的电子密度差异如图5b所示。结果表明异质界面和单个Fe原子位置在吸附H2O分子时表现出明显的电子损耗,证明有效促进了电子从活性位置转移到H2O分子。
为了检验H*吸附/H2解吸步骤(Heyrovsky步骤),计算了所有可能的活性位点的吉布斯自由能(ΔGH*)(图5c),相应的原子构型显示在图5d中。图5c显示,单相模型中所有活性位点上H*吸附的ΔGH*值超出0.3–0.3 eV的范围,表明HER性能较差。而异质结构界面和单原子分散模型中大多数活性位点的ΔGH*值在0.19–0.22 eV范围内(MoP/MoP2除外)。特别地,MoP2/MoO2中的
为了阐明H*吸附的物理机制,通过研究H*吸附在异质界面和单原子分散模型的表面位点上产生的二维电子密度差异(图5e),分析了H*和活性位点之间电子转移的大小。在Fe单原子分散模型中,发现Fe@MoO2-1中Fe位点的电子转移比Fe@MoO2-2中的大。这与它们的ΔGH*值的差异相一致(Fe@MoO2-1为-0.19 eV,Fe@MoO2-2为0.16 eV)。有利的ΔGH*值表明,在MoO2中与一个或两个O原子结合的单原子分散的铁原子具有适当的H*吸附/解吸能力,可以有效地进行HER电催化。
总结与展望
本文通过对环境友好自组装有机无机配位的FePMoG纳米片进行低温磷化制备了一种由多孔的Fe/SAs@Mo基HNSs组成的高效HER电催化剂。电化学测试表明此催化剂具有迄今为止报道的纳米结构的Mo基电催化剂的最佳HER性能之一。这种出色的性能归因于Fe/SAs@Mo基HNSs优化的电子结构、丰富的界面和边界相、较大的活性表面积和孔隙率,以及单原子和异质结构的协同效应。这种高性能的无碳电催化剂是易腐蚀的含碳催化剂的有效替代品,可用于质子交换膜燃料电池和其他先进的能源技术。
审核编辑:郭婷
-
国仪电镜助力PANC/T-Fe水凝胶在不同环境温度下的微观结构分析2025-07-30 6362
-
碳纳米纤维的应用前景怎么样?2019-09-20 3308
-
铁的原子结构示意图2008-05-28 125322
-
水凝胶的介绍和生物医用高强度水凝胶的力学强度分析2017-09-19 2450
-
纳米技术对镁碳耐火材料的改进2018-02-11 1484
-
科学家研发可坚持数月的新型水凝胶2021-02-05 2317
-
简述石墨烯纳米结构的原子级精准构造2021-06-17 4934
-
一种纳米纤维水凝胶(EKGel)用于乳腺癌PDOs的形成和生长2022-07-10 2110
-
具有极端力学行为的水凝胶3D打印设计与制造2022-07-11 2192
-
科学家开发石墨烯气凝胶颗粒,用于高效水净化2023-02-16 1869
-
用于极端环境下的热防护材料——仿贝壳纳米复合气凝胶2023-06-27 2060
-
基于结构色水凝胶的生物传感器应用2023-11-20 1987
-
水凝胶中的可控刺激响应行为2023-11-28 940
-
石墨烯异质结构新进展2025-02-05 1203
-
水凝胶拉伸试验机:材料性能的洞察者2025-04-28 710
全部0条评论

快来发表一下你的评论吧 !
