

具有高硫载量和高效转化动力学开发RT-Na/S电池应用
描述
01、导读
由于钠和硫的丰富性和高理论容量,室温钠硫(RT-Na/S)电池是最有前景的低成本和高能量密度系统之一。然而,RT-Na/S电池面临着一些挑战,例如硫的绝缘性和快速的容量衰减。此外正极中硫活性物质的低载量仍不能满足实际应用的要求。因此,合理设计具有高硫载量和高效转化动力学的S主体是开发实用的RT-Na/S电池的关键。
02、成果背景
为了应对上述RT-Na/S的挑战,人们提出了诸多解决方法。其中,具有双活性位点的金属硫化物基电催化剂因其在捕获多硫化物以转化为Na2S和减轻“穿梭效应”方面的优势而受到越来越多的关注。在2019年由多个团队陆续提出因原子级分散的双中心催化剂其独特的电子结构、活性原子中心和低配位环境成为高性能硫正极的理想候选材料(Small Methods 2019, 3, 1800497;Nat. Catal. 2019, 2, 304. D),此后该类型催化剂进一步发展。
近日Adv. Mater. 期刊上发表了一篇题为“Atomically Dispersed Dual-Site Cathode with a Record High Sulfur Mass Loading for High-Performance Room-Temperature Sodium-Sulfur Batteries”的文章。该工作合成了支持原子级分散的2H-MoS2和Mo1(S@MoS2-Mo1/SGF)的硫掺杂石墨烯骨架,其硫载量达到创纪录的80.9 wt%,用其作为RT-Na/S电池的集成双活性位点正极。制备的S@MoS2-Mo1/SGF显示了极佳的循环稳定性,在0.1 A g-1时具有1017 mAh g-1的高初始容量和超过1000次循环时0.05%的低容量衰减率。X射线吸收光谱(XAS)、原位同步X射线衍射(XRD)和密度泛函理论计算等结果表明,该集成双活性位的原子级Mo形成了离域电子体系,提高了硫的反应活性和S、Na的反应可逆性,极大地缓解了穿梭效应。这些发现不仅为制备高性能双位点正极提供了有效的策略,而且加深了对其增强机制在原子水平上的理解。
03、关键创新
(1)合成了硫掺杂的石墨烯骨架,RT-Na/S电池的S载量高达80.9%,具有高初始容量和低容量衰减率;
(2)将原位X射线技术与计算相结合研究了原子级双活性位正极的增强机制,表明原子级分散的双位点系统可以产生离域电子效应,优化活性钼的电子结构,导致钠中间体的吸附能为负,还有利于促进多硫化钠的转化动力学,从而抑制穿梭效应。
04、核心内容解读
(1)环绕在硫掺杂石墨烯框架上的原子级双活性位点
S@MoS2-Mo1/SGF的透射电子显微镜(TEM)图像(图1a)表明石墨烯骨架的结构在负载硫后得到很好的保持。采用高角度环形暗场(HAADF)扫描透射电子显微镜(STEM)确认Mo1/SGF和S@Mo1/SGF表面没有明显的纳米团簇或纳米颗粒。在S@MoS2-Mo1/SGF的HAADF-STEM图像中可以观察到MoS2单层纳米团簇和锚定在SGF表面的单个Mo原子(图1b)。元素映射图像(图1c)表明,得到的S@MoS2-Mo1/SGF不含其他杂质,C、S和Mo在整个表面上均匀分散。为了研究MoS2单层结构,采用了高分辨率STEM(HR-STEM)图像和定量STEM(QSTEM)模拟(图1d和1e)。HAADF-STEM图像(图1d)显示,这些簇在石墨烯上的原子结构是沿[001]区轴的典型六边形结构。通过QSTEM软件模拟原子分辨率HAADF-STEM图像(图1e)来识别S@MoS2-Mo1/SGF模型(图1f)。参与反应的Mo和S均匀分布在石墨烯表面,并以单层六方MoS2的形式生长。这些实验结果与模拟相结合,证实成功合成了S@MoS2-Mo1/SGF。
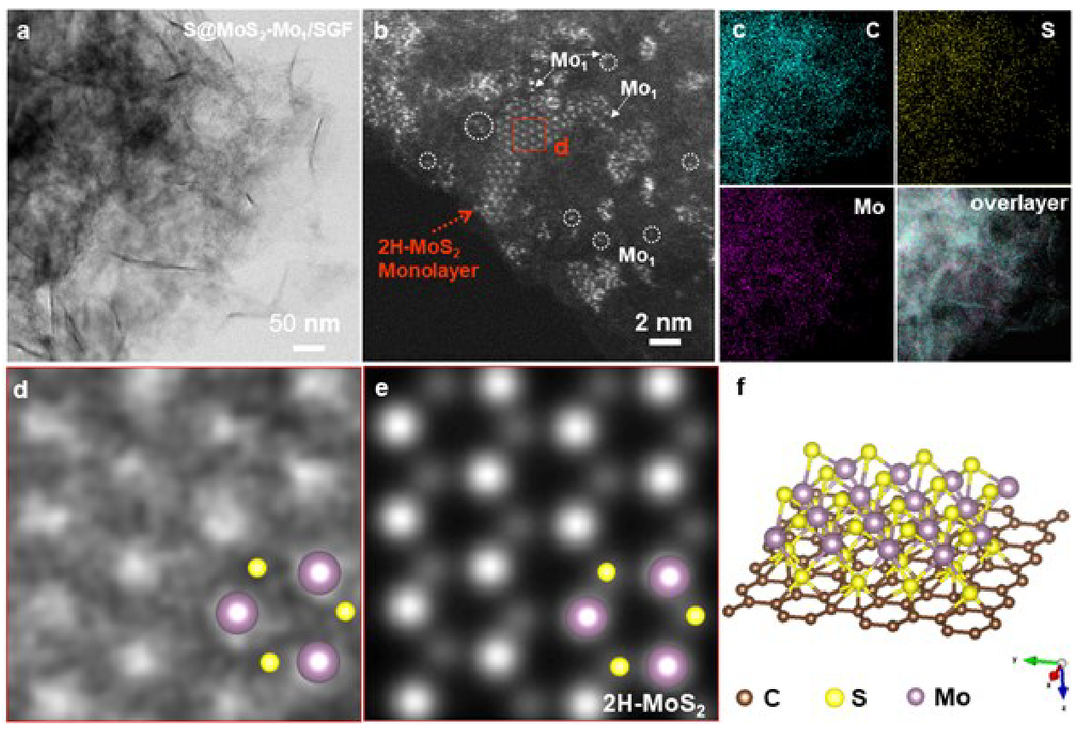
图1 电子显微镜图像。a-c、TEM、HAADF-STEM图像和S@MoS2-Mo1/SGF的相应元素映射图像。d-f,HR-STEM视图和QSTEM模型对应于2H-MoS2单层结构的示意图。
X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)测试(图2a、2b)用于研究S@MoS2-Mo1/SGF的配位环境和电子结构。较高的Mo化学价可归因于双位点MoS2-Mo1体系的形成,这有助于电子从Mo转移到负载的S。Mo1/SGF和S@MoS2Mo1/SGF的EXAFS结果(图2b)表明Mo1/SGF具有一个大约1.1 Å的主要间距,而在2.4 Å处不存在Mo-Mo可能是由于存在单个Mo原子。如图1b所示,单层MoS2簇被单个Mo原子包围。这种短程在单层MoS2和Mo1之间产生离域电子界面,导致电子从Mo转移到S。这就是S@MoS2-Mo1/SGF中Mo的化学价高于S@MoS2/SGF的原因。EXAFS结果表明,经过较长时间的热处理,单个Mo原子可以更完全地组装成MoS2。
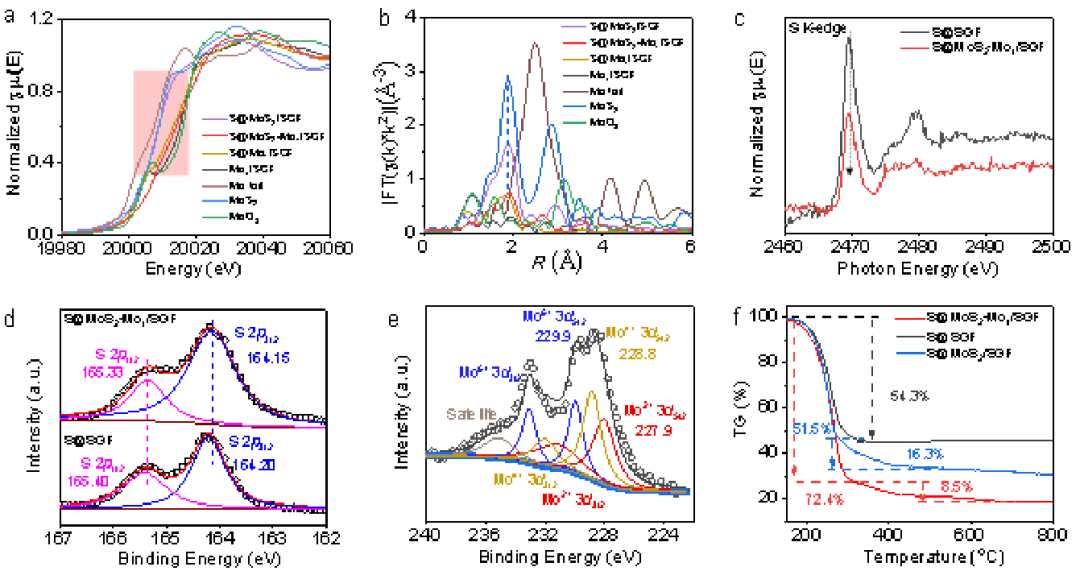
图2 S@MoS2-Mo1/SGF的表征。a,Mo K边XANES光谱和b,S@MoS2-Mo1/SGF、S@MoS2/SGF、S@Mo1/SGF、Mo1/SGF、Mo箔、MoS2和MoO3的R-space EXAFS光谱。c,S K边NEXAFS光谱,d和e,S@MoS2、Mo1/SGF和S@SGF的XPS结果。f,S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF的TGA结果。
收集S@MoS2-Mo1/SGF和S@SGF的近边X射线吸收精细结构(NEXAFS)光谱(图2c)以研究硫的电子转移,验证了从MoS2-Mo1/SGF到S的电子转移。转移电子将激活硫并增强其反应性和多硫化物形成的动力学。X射线光电子能谱(XPS)用于进一步研究样品中的化学状态(图2d、2e)。图2d显示了S@MoS2-Mo1/SGF(图2d)的S 2p3/2峰(164.15 eV)与S@SGF(164.20 eV)相比负移了0.05 eV,这表明S是电子受体。图2e中的XPS光谱表明Mo的化学价由+2、+4和+6组成。在S@MoS2-Mo1/SGF的光谱中可以观察到各种Mo2+ 3d5/2、Mo4+ 3d5/2和Mo6+ 3d5/2子光谱,这归因于MoS2和原子Mo1之间的离域电子效应。此外,与纯MoS2相比,S@MoS2-Mo1/SGF的Mo4+ 3d5/2显示出0.8 eV的左移,这表明电子从离域电子系统转移到S。热重分析(TGA)测试结果(图2f)表明,S@MoS2-Mo1/SGF介孔中储存的S含量为80.9 wt%,高于S@MoS2/SGF(67.8 wt%)和S@SGF(54.3 wt%)。
(2)室温钠硫电池的电化学性能
S@MoS2Mo1/SGF、S@MoS2/SGF、S@Mo1/SGF和S@SGF在0.1 A g-1下的第1、2、5和10次循环的放电/充电曲线如图3a、3b所示。S@Mo1/SGF的首圈放电容量高于S@SGF(522 mAh g-1),表明原子级分散的Mo可以提高电化学性能。
图3c显示S@MoS2-Mo1/SGF在电流密度分别为0.2、0.5、1、2和5 A g-1时具有1042、831、529、281和171 mAh g-1的最佳可逆容量(图3c)。图3d显示了S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF电极的循环稳定性。S@MoS2-Mo1/SGF正极在1000次循环后仍可保持505 mAh g-1的容量,每循环0.05%的容量衰减率极低。S@MoS2-Mo1/SGF的超低容量衰减率归因于在原子级分散的MoS2-Mo1中构建的电子离域界面,该界面可以提供自由电子将多硫化物快速还原成最终产物Na2S。S@MoS2Mo1/SGF正极也在高电流密度(图3c)和高硫载量下进行了研究,这通常需要更快的多硫化物转化的氧化还原动力学。在所有报道的文章中,S@MoS2-Mo1/SGF表现出最佳的循环性能和最高的硫容量(图3g)。为了研究多硫化物和Na2S的动力学转化,对S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF在不同扫描速率(v)下的循环伏安图(CV)进行了分析。图3e展示出S@MoS2-Mo1/SGF的DNa+最大,表明多硫化钠向Na2S的动力学转化最快。
进行原位同步辐射XRD(λ=0.5904 Å)以揭示S@MoS2-Mo1/SGF正极的储钠机制(图3f)。放电过程如以下方程(1)-(2)所示
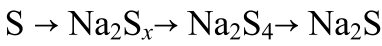 (1)
(1)
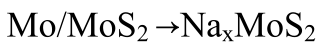 (2)
(2)
充电过程可描述为:
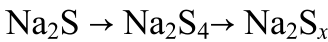 (3)
(3)
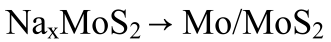 (4)
(4)
因此,从Na2S到Na2S4的转化过程是快速且高度可逆的,从而导致优异的循环性能和倍率性能。还通过将Na2S6暴露于MoS2-Mo1/SGF和SGF来研究这些材料对多硫化物的化学吸附性能,表明MoS2-Mo1/SGF表现出对Na2S6的有效限制以及将Na2S6电催化转化为最终Na2S的出色性能。
图3 RT-Na/S电池性能。a、S@MoS2-Mo1/SGF和b、S@SGF在100 mA g-1下的放电/充电曲线。c,倍率性能,d,循环性能,e,S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF的Na+离子扩散系数。f,S@MoS2-Mo1/SGF正极的原位同步辐射XRD,对应于0.5 A g-1下的初始恒电流充放电曲线。g,S@MoS2-Mo1/SGF正极与之前报道的文献之间的电池性能比较。
(3)RT-Na/S电池中硫正极的机理研究
进行密度泛函计算以研究MoS2-Mo1/SGF优异电化学催化性能的来源,如图4a所示。
与S@MoS2/SGF相比,S@MoS2-Mo1/SGF上原子级分散的MoS2Mo1界面伴随着明显的电荷转移。从S@MoS2Mo1/SGF和S@MoS2/SGF的相应计算的部分状态密度(PDOS)获得的d带中心(Ed)如图4b所示,与S@Mo1/SGF(-0.73 eV)相比,S@MoS2-Mo1/SGF的Mo单原子的Ed值变为-0.41 eV,表明催化活性增强。
图4 机制研究。a,Mo中4d电子和S元素中3p电子以不同价数配位的分子轨道示意图。b,d带集中在S@MoS2-Mo1/SGF和S@MoS2/SGF的相应活性Mo位点上。c,在S@MoS2Mo1/SGF、S@MoS2/SGF和SGF上从Na2S4到Na2S中间体放电过程的能量变化图。d,在相应的钠硫电池模型上,Na2S4的吸附能(eV)作为从头算分子动力学(AIMD)模拟时间的函数。
接下来,基于三个模型计算了从Na2S4到Na2S中间体的能量变化图,以模拟RT-Na/S电池的放电过程(图4c),表明放电过程不能通过第一个中间体Na2S4。
为了使计算更接近于室温下的现有系统,进行了从头算分子动力学(AIMD)模拟(图4d)。经过5 ps的动力学模拟,发现S@MoS2-Mo1/SGF在三种模型中唯一具有较大的负吸附能(约-12 eV),与热力学结果一致。相反,S@MoS2/SGF和SGF会遇到巨大的正能量势垒(约6 eV)来完成放电过程。还注意到S@MoS2-Mo1/SGF的最终吸附态结构更倾向于Na2S中间体,而S@MoS2/SGF和SGF上的最终吸附态结构分别接近Na2S3和Na2S4。
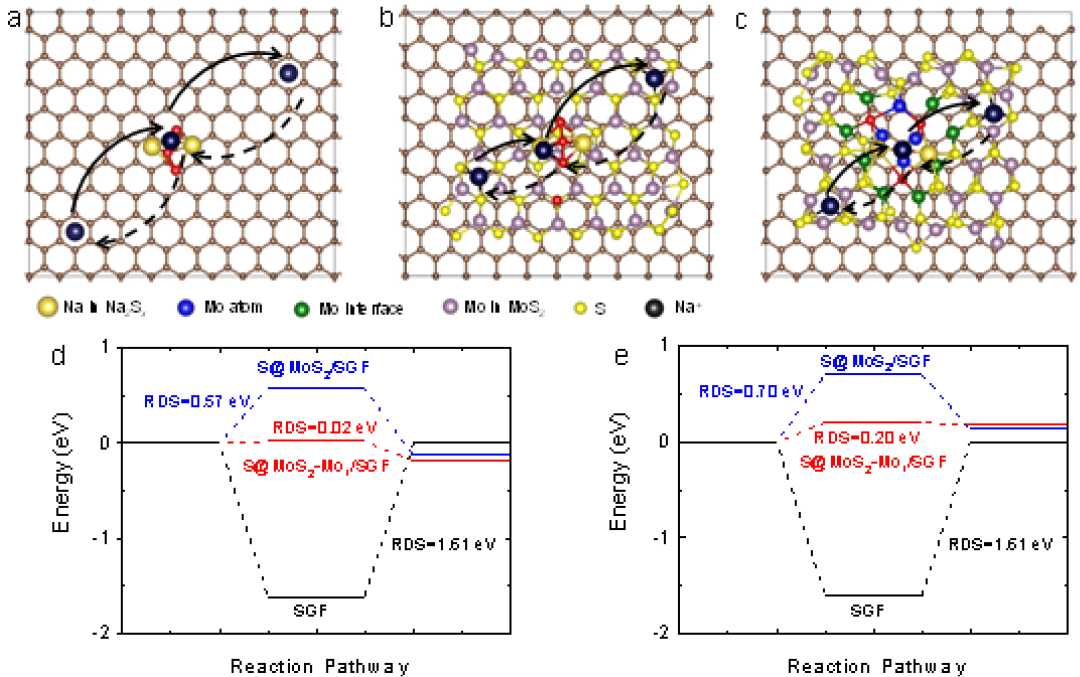
图5钠离子在硫宿主上迁移。a,Na2S4*SGF,b,Na2S3*S@MoS2/SGF,c,Na2S1*S@MoS2-Mo1/SGF中的钠离子迁移途径。硫的数量来自先前热力学计算中最稳定的吸附物,用于相应结构计算的钠离子迁移势垒在(d)固体和(e)虚线方向上的能垒。
此外,通过计算分析研究了钠离子在三个模型上的迁移(图5a-5c)。计算了钠离子在不同位置的形成能。选择了最大可能的反应路径,如图5d和图5e所示。根据阿伦尼乌斯定理,这些结果表明钠离子的迁移速率顺序为:MoS2Mo1/SGF>S@MoS2/SGF>SGF,这与实验观察结果一致。
05、成果启示
具有原子级双活性位点的S@MoS2-Mo1/SGF材料被合成作为RT-Na/S电池正极的优质硫主体。原子级分散的双活性位点以其独特的配位环境产生离域电子,可为硫提供自由电子,从而有效提高硫的反应性和多硫化物的动力学转化。S@MoS2-Mo1/SGF材料在1000次循环后表现出505 mAh g-1可逆容量,每个循环的容量衰减率低至0.05%。一系列的实验表征和计算表明,S@MoS2Mo1/SGF的优异性能可归因于创建的层状MoS2-Mo1位点,这可优化中间体的吸附能并将多硫化物自发分解为Na2S。
审核编辑:郭婷
-
Adams多体动力学仿真解决方案全面解析2025-04-17 7778
-
轮毂电机驱动电动汽车垂向动力学控制研究综述2025-03-07 418
-
基于车辆动力学模型的横向控制2023-11-15 1840
-
V2C MXene组件促进实用锂硫电池的硫释放动力学和锂离子筛分2023-04-01 2320
-
分布式驱动电动汽车的动力学控制有哪几种类型?常见问题是什么?2021-08-30 1297
-
飞行器动力学参数在线辨识EKF算法实验流程2021-08-27 1370
-
基于dSPACE的嵌入式车辆动力学仿真平台开发的探究2021-07-30 820
-
电动力学或经典电动力学统称为什么?2020-06-10 4551
-
汽车系统动力学长篇大论2017-11-01 4982
-
高梯度磁场中磁性微粒的动力学模型研究2010-05-13 2184
-
基于多体系统动力学的空气悬架大客车平顺性试验仿真研究2009-12-02 4065
-
热分析动力学2009-12-01 681
-
[下载]想了解多体动力学软件吗?有教程分享及免费试用下载2009-03-24 4228
-
电力拖动系统的动力学课件2008-11-19 4382
全部0条评论

快来发表一下你的评论吧 !
