

利用Amber热力学积分计算相对自由能变化
电子说
描述
上周四,何博士为大家在北鲲云的直播间分享了Amber热力学积分计算相对自由能变化)。
直播结束后有很多小伙伴来向我们要PPT资料,这里何博士也为大家准备了文字版本的教程。将为大家介绍如何在北鲲云计算平台上利用Amber热力学积分计算相对自由能变化,体系包括小分子-蛋白(小分子改变),小分子-蛋白(蛋白突变),蛋白-蛋白相互作用。
本教程要求使用者一定程度了解Amber动力学模拟程序。
Amber是美国加州大学Peter Kollman等开发的一款著名的分子动力学模拟软件包。Amber主要适用于蛋白质,小分子和多糖等生物分子体系的模拟。
本文所需的所有文件请在https://github.com/Xinheng-He/ti_toturial上下载。

该应用场景解决将蛋白口袋内的小分子A变为小分子B所产生的相对自由能变。
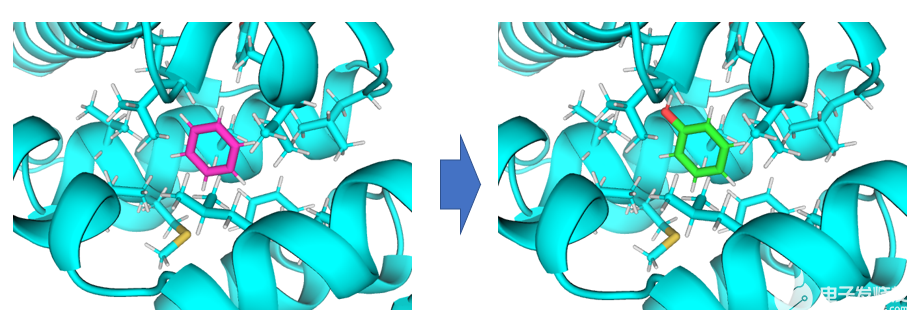
将蛋白口袋内的苯转化为苯酚。
首先,使用pymol将分子打开,并选中小分子,保存为mol2文件,如下图所示,我们使用
save *my_caseben_ligand.mol2save *my_casebenfen_ligand.mol2
这两个命令保存了变化前后的配体。
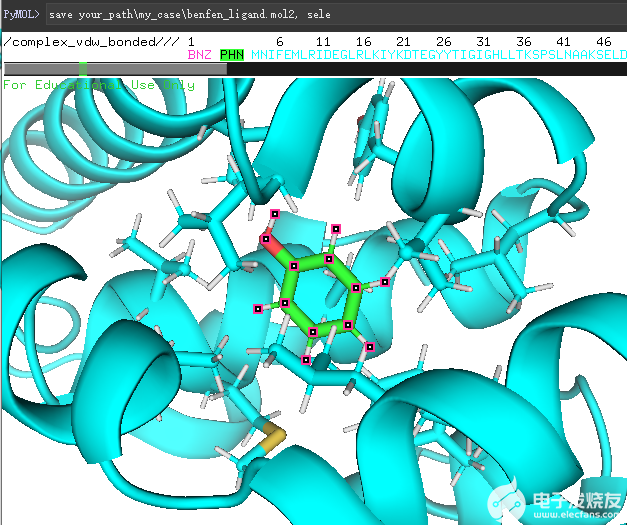
(保存pymol中的sele对象)
开启一个北鲲云管理节点加载环境。
module add Anaconda3/2020.02 source /public/software/.local/easybuild/software/amber/aber20/amber.sh ulimit -s unlimited ulimit -l unlimited
对苯生成具有电荷的可用mol2文件,总电荷为0,残基名为BEN。
antechamber -i ben_ligand.mol2 -fi mol2 -o ben_real.gaff2.mol2 -fo mol2 -rn BEN -at gaff2 -an yes -dr no -pf yes -c bcc -nc 0
生成frcmod力场参数文件。
parmchk2-iben_real.gaff2.mol2-fmol2-oBEN.gaff2.frcmod-sgaff2-ayes
上述操作对苯酚再来一次。
antechamber -i benfen_ligand.mol2 -fi mol2 -o benfen_real.gaff2.mol2 -fo mol2 -rn FEN -at gaff2 -an yes -dr no -pf yes -c bcc -nc 0 parmchk2 -i benfen_real.gaff2.mol2 -f mol2 -o FEN.gaff2.frcmod -s gaff2 -a yes
根据frcmod文件,生成两个分子的文库文件,该文件描述了分子内部的原子类型和键连信息。
tleap -f - <<_EOF source leaprc.gaff2 loadamberparams FEN.gaff2.frcmod FEN = loadmol2 benfen_real.gaff2.mol2 saveoff FEN FEN.lib savepdb FEN FEN.pdb quit _EOF tleap -f - <<_EOF source leaprc.gaff2 loadamberparams BEN.gaff2.frcmod BEN = loadmol2 ben_real.gaff2.mol2 saveoff BEN BEN.lib savepdb BEN BEN.pdb quit _EOF
注意前后的力场要保持一致。
将两个pdb文件(FEN.pdb和BEN.pdb)中同样位置的全部重原子,保存成同样的坐标,注意名字要和lib中的一样,放成一个lig.pdb,在下面的tleap过程中,tleap会自动根据lib文件将complex中的原子变成真实的样子,这样做是为了保证一样原子的位置完全一致,减少不必要的变量。
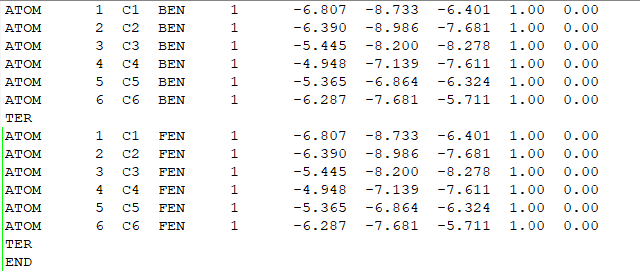
(pdb文件的内容)
使用pdb4amber,检查蛋白是否有二硫键,或需要编辑的残基。
pdb4amber pure_protein.pdb -o pure_check.pdb cat pure_check_sslink
没有二硫键,之后使用pure_check.pdb。
接下来在tleap中加载配体和受体。
tleap -f - <<_EOF
source leaprc.protein.ff14SB
source leaprc.gaff2
source leaprc.water.tip3p
loadAmberParams frcmod.ionsjc_tip3p
loadoff BEN.lib
loadoff FEN.lib
loadamberparams BEN.gaff2.frcmod
loadamberparams FEN.gaff2.frcmod
ligands = loadpdb lig.pdb
complex = loadpdb pure_check.pdb
complex = combine {ligands complex}
check complex
solvatebox ligands TIP3PBOX 15
addions ligands Na+ 0
savepdb ligands ligands_vdw_bonded.pdb
saveamberparm ligands ligands_vdw_bonded.parm7 ligands_vdw_bonded.rst7
solvatebox complex TIP3PBOX 15
addions complex Cl- 0
savepdb complex complex_vdw_bonded.pdb
saveamberparm complex complex_vdw_bonded.parm7 complex_vdw_bonded.rst7
quit
_EOF
注意根据实际电荷情况调整addions,如果ligand/complex带负电,加Na+,反之加Cl-,离子类型可以自己选择。
下载complex_vdw_bonded.pdb并检查其中的配体分子区别,是否只是不一样的配体部分不一样,确定配体不一样部分的原子。设置出发和结束原子。
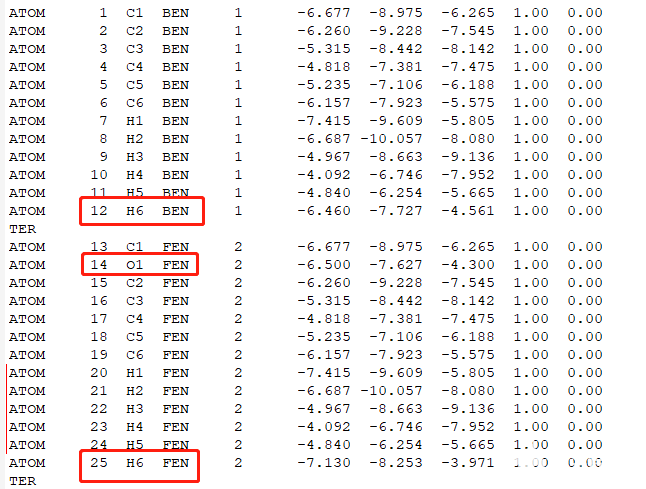
红框是有区别的原子,其他原子需要手动编辑使其保持一致的坐标,红线是编辑后的原子。
修改initial中的in文件下述部分。
timask1 = ':BEN', timask2 = ':FEN', scmask1 = ':BEN@H6', scmask2 = ':FEN@O1,H6'
使6个in文件符合实际更改的原子情况,只改变不同的原子,该步骤的目标是优化vdw变化过程中氢原子的位置。
使用sbatch run_v100.slurm,将任务提交到北鲲云的单卡V100集群上,该任务大概耗时1分钟,注意该任务如果有报错,可能是以下问题:
如果上述过程报关于ambmask的错(比如mask中不能用_符号),可以改写ambmask来获得正确可识别的mask。
ambmask -p complex_vdw_bonded.parm7 -c complex_vdw_bonded.rst7 -find :FEN
是ambmask的输入方式,在parm7和rst7中寻找这些原子,并用pdb的形式输出。
如果报错Error : Atom 12 does not have match in V1 ,说明这个原子在两个小分子配体中间的位置区别太大,TI不能识别这两个分子作为一样的背景,因此将这两个原子(初始和结束配体的)加入坐标中,就可以解决这个问题。
运行结束后,提取优化过后的分子结构。
p ligands_vdw_bonded.rst7 ligands_vdw_bonded.rst7.leap cp press_lig.rst7 ligands_vdw_bonded.rst7 cp complex_vdw_bonded.rst7 complex_vdw_bonded.rst7.leap cp press_com.rst7 complex_vdw_bonded.rst7 cpptraj -p ligands_vdw_bonded.parm7 <<_EOF trajin ligands_vdw_bonded.rst7 strip ":1,2" outtraj ligands_solvated.pdb onlyframes 1 unstrip strip ":2-999999" outtraj ligands_BEN.pdb onlyframes 1 unstrip strip ":1,3-999999" outtraj ligands_FEN.pdb onlyframes 1 _EOF cpptraj -p complex_vdw_bonded.parm7 <<_EOF trajin complex_vdw_bonded.rst7 strip ":1,2" outtraj complex_solvated.pdb onlyframes 1 unstrip strip ":2-999999" outtraj complex_BEN.pdb onlyframes 1 unstrip strip ":1,3-999999" outtraj complex_FEN.pdb onlyframes 1 _EOF
再次使用tleap生成decharge,vdw和recharge的文件,decharge是配体1,recharge是配体2,修改阅读的pdb的名字即可。
tleap -f - <<_EOF
# load the AMBER force fields
source leaprc.protein.ff19SB
source leaprc.gaff2
source leaprc.water.tip3p
loadAmberParams frcmod.ionsjc_tip3p
loadOff BEN.lib
loadOff FEN.lib
loadamberparams BEN.gaff2.frcmod
loadamberparams FEN.gaff2.frcmod
# coordinates for solvated ligands as created previously by MD
lsolv = loadpdb ligands_solvated.pdb
lbnz = loadpdb ligands_BEN.pdb
lphn = loadpdb ligands_FEN.pdb
# coordinates for complex as created previously by MD
csolv = loadpdb complex_solvated.pdb
cbnz = loadpdb complex_BEN.pdb
cphn = loadpdb complex_FEN.pdb
# decharge transformation
decharge = combine {lbnz lbnz lsolv}
setbox decharge vdw
savepdb decharge ligands_decharge.pdb
saveamberparm decharge ligands_decharge.parm7 ligands_decharge.rst7
decharge = combine {cbnz cbnz csolv}
setbox decharge vdw
savepdb decharge complex_decharge.pdb
saveamberparm decharge complex_decharge.parm7 complex_decharge.rst7
# recharge transformation
recharge = combine {lphn lphn lsolv}
setbox recharge vdw
savepdb recharge ligands_recharge.pdb
saveamberparm recharge ligands_recharge.parm7 ligands_recharge.rst7
recharge = combine {cphn cphn csolv}
setbox recharge vdw
savepdb recharge complex_recharge.pdb
saveamberparm recharge complex_recharge.parm7 complex_recharge.rst7
quit
_EOF
生成好这些过程的文件后,使用setup_run.sh来产生三个步骤的输入文件,在修改setup_run.sh时,注意以下部分。
decharge_crg=":2@H6" vdw_crg=":1@H6 | :2@O1,H6" recharge_crg=":1@O1,H6" decharge=" ifsc = 0, crgmask = '$decharge_crg'," vdw_bonded=" ifsc=1, scmask1=':1@H6', scmask2=':2@O1,H6', crgmask='$vdw_crg'" recharge=" ifsc = 0, crgmask = '$recharge_crg',"
适配修改,注意H6的都改成初始的ambmask,O1,H6的都改成目标的ambmask,:前面的1或者2不要改。
如有必要修改λ,改变prod.tmpl和setup.sh中的值(0.00922开始的那一串)。
该文件将直接生成所需的slurm文件,并提交到对应的机器上,默认使用g-v100-1,运行pmemd.cuda,大致运行时间1小时左右。
有时候pmemd.cuda会运行失败,此时转用cpu来运行,使用run_mpi.py,提交命令:
python run_mpi.py ligands 500000 mpi
将会检查所有ligands下的文件,对于5分钟内没更新,且info中运行步骤在500000 以下的,会提交到32核CPU机器上运行后续的模拟,直到结束。
这个运行步骤非常缓慢,使用32核CPU算完1纳秒的步骤可能需要7-8天,可以尝试先运行一段CPU,再运行一段GPU,改为python run_mpi.py ligands 500000 cuda即可。
运行结束后,使用alchemical-analysis/alchemical_analysis/alchemical_analysis.py来分析结果,注意该文件在python2下运行。
pip2 install matplotlib pip2 install scipy pip2 install numpy pip2 install pymbar==3.0.3
运行:
mkdir -p ana_recharge && cd ana_recharge ../../alchemical-analysis/alchemical_analysis/alchemical_analysis.py -a AMBER -d . -p ../recharge/[01]*/ti00[1-9] -q out -o . -t 300 -v -r 5 -u kcal -f 50 -g -w mkdir -p ../ana_decharge && cd ../ana_decharge ../../alchemical-analysis/alchemical_analysis/alchemical_analysis.py -a AMBER -d . -p ../decharge/[01]*/ti00[1-9] -q out -o . -t 300 -v -r 5 -u kcal -f 50 -g -w mkdir -p ../ana_vdw && cd ../ana_vdw ../../alchemical-analysis/alchemical_analysis/alchemical_analysis.py -a AMBER -d . -p ../vdw_bonded/[01]*/ti00[1-9] -q out -o . -t 300 -v -r 5 -u kcal -f 50 -g -w
此时只能输出三种变化的结果,将其TI 一列加和得到最终的结果。
如果见到pymbar的warning,只要注释掉对应的assert就可以了。
vim /home/cloudam/.local/lib/python2.7/site-packages/pymbar/timeseries.py
去第162行。

该应用场景解决将蛋白口袋内的残基A变为残基B所产生的相对自由能变。
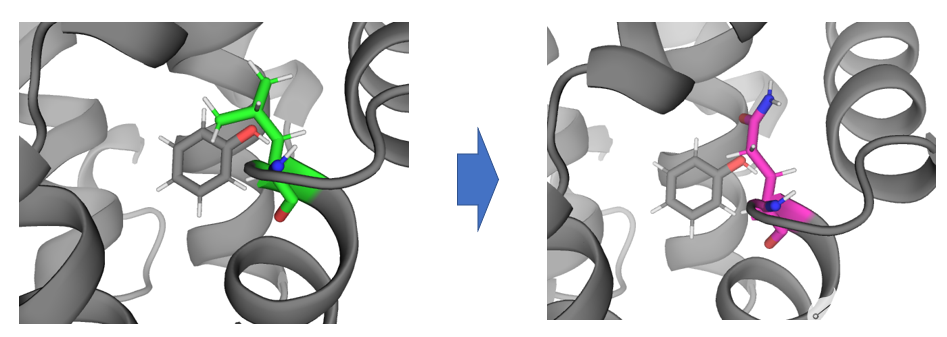
将蛋白口袋内的LEU转化GLN。
首先,使用pymol将分子打开,并选中残基,使用wizard-mutagenesis-protein完成突变,或者使用命令行完成突变:
load *.pdb
cmd.wizard("mutagenesis")
cmd.do("refresh_wizard")
cmd.get_wizard().set_mode("GLN")
cmd.get_wizard().do_select("86/")
cmd.get_wizard().apply()
cmd.set_wizard("done")
save *out_name.pdb, enabled
将突变后的蛋白文件保存为pdb文件。
将突变前的蛋白(WT.pdb)和突变后的蛋白(L86Q.pdb)的pdb文件上传。使用tleap,读取野生型结构。
tleap source leaprc.protein.ff14SB source leaprc.gaff2 source leaprc.water.tip3p loadamberparams frcmod.ionsjc_tip3p zn = loadpdb WT.pdb check zn solvateBox zn TIP3PBOX 10 addIons zn Cl- 0 savepdb zn box_check.pdb quit
记录盒子的范德华半径,并将结构中的水提取出来备用。
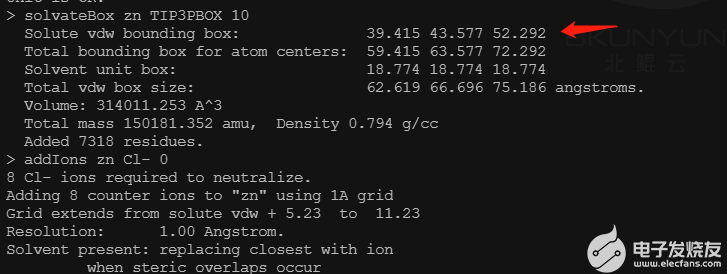
python dry_for_TI.py box_check.pdb wat.pdb WT_receptor.pdb
同样的方式读取突变型结构,使其保持Amber的原子顺序。
tleap source leaprc.protein.ff14SB source leaprc.gaff2 source leaprc.water.tip3p loadamberparams frcmod.ionsjc_tip3p zn = loadpdb L86Q.pdb check zn solvateBox zn TIP3PBOX 10 addIons zn Cl- 0 savepdb zn L86Q_leap.pdb quit python dry_for_TI.py L86Q_leap.pdb wat1.pdb L86Q_dry.pdb
对比两个去水后的文件,发现由于Amber重编号,突变的残基变为84位,此时使用check_diff_online.py,来保证不是突变的残基的位置都绝对一致,以允许进行TI过程。
python check_diff_online.py L84Q L86Q_dry.pdb WT_receptor.pdb 84 L86Q_check.pdb WT_check.pdb
print的是空字典,说明所有的原子都是匹配的。
再次用tleap读取受体和配体(之前第一部分保存的mol2文件),并读取水盒子,其中ligand是刚才保存的配体(不变部分),m1和m2分别是突变前后的部分(注意突变只改变侧链,不改变主链),注意这里使用了刚才记录的范德华半径。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff2
loadOff FEN.lib
loadamberparams FEN.gaff2.frcmod
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
ligand = loadmol2 FEN.gaff2.mol2
m1 = loadpdb L86Q_check.pdb
m2 = loadpdb WT_check.pdb
w = loadpdb wat1.pdb
protein = combine {m1 m2 w}
complex = combine {m1 m2 ligand w}
set default nocenter on
setBox protein vdw {39.415 43.577 52.292}
savepdb protein protein.pdb
saveamberparm protein protein.parm7 protein.rst7
setBox complex vdw {39.415 43.577 52.292}
savepdb complex complex.pdb
saveamberparm complex complex.parm7 complex.rst7
quit
使用parmed处理protein.parm7和complex.parm7,以保证正确的所改变的位置提供给TI运算,此处的162为WT或者突变体的残基数,@后面的内容是python get_mutation.py L86Q得到的mapping结果,是在那个突变残基上但也没有变化的残基,84是突变位置,246是84+162。
parmed protein.parm7 <<_EOF loadRestrt protein.rst7 setOverwrite True tiMerge :1-162 :163-324 :84&!@CA,C,O,N,H,HA,CB :246&!@CA,C,O,N,H,HA,CB outparm merged_L86Q_protein.parm7 merged_L86Q_protein.rst7 quit _EOF parmed complex.parm7 <<_EOF loadRestrt complex.rst7 setOverwrite True tiMerge :1-162 :163-324 :84&!@CA,C,O,N,H,HA,CB :246&!@CA,C,O,N,H,HA,CB outparm merged_L86Q_complex.parm7 merged_L86Q_complex.rst7 quit _EOF
正确获得这些文件后,使用:
python auto_gene_inp_run.py 84 162 CA,C,O,N,H,HA,CB L86Q
来生成tmpl文件(run.tmpl文件需要上传),并将tmpl文件转成真正的ti文件放进文件夹,同时使用slurm提交最小化,加热和运行步骤。
注意,这里直接使用cuda很容易断,可以适当自己修改之前的run_mpi.py来使用cpu续跑中断的模拟。
运行结束后的分析略。

本案例将实际运行一个蛋白-蛋白相互作用上的突变。我们计算新冠病毒受体结合区域(rbd)到人ACE2受体(ace2)复合物上发生Omicron的突变之一的返回突变A484E后的结合自由能变化。
生成连接了二硫键的大复合体水盒,记录vdw盒子大小,额外加入0.15M/L NaCL (总水数量*0.002772)。
tleap source leaprc.protein.ff14SB source leaprc.gaff source leaprc.water.tip3p loadamberparams frcmod.ionsjc_tip3p zn = loadpdb omi_SS.pdb bond zn.333.SG zn.358.SG bond zn.376.SG zn.429.SG bond zn.388.SG zn.522.SG bond zn.477.SG zn.485.SG bond zn.637.SG zn.645.SG bond zn.848.SG zn.865.SG bond zn.1034.SG zn.1046.SG check zn solvateBox zn TIP3PBOX 10 addIons zn Na+ 0 addIons zn Na+ 80 addIons zn Cl- 0 savepdb zn box_check.pdb quit python dry_for_TI.py box_check.pdb wat1.pdb omi_rbd.pdb,omi_ace2.pdb
同时生成一个小的水盒,用于跑蛋白部分的TI。
tleap source leaprc.protein.ff14SB source leaprc.gaff source leaprc.water.tip3p loadamberparams frcmod.ionsjc_tip3p m1 = loadpdb omi_rbd.pdb bond m1.4.SG m1.29.SG bond m1.47.SG m1.100.SG bond m1.59.SG m1.193.SG bond m1.148.SG m1.156.SG check m1 solvateBox m1 TIP3PBOX 10 addIons m1 Na+ 0 addIons m1 Na+ 28 addIons m1 Cl- 0 savepdb m1 ligands_recharge.pdb quit python dry_for_TI.py ligands_recharge.pdb rbd_wat.pdb test_rbd.pdb
检查输入的是否在TI区域之外没有区别,注意输入的顺序,前面是突变后的蛋白,后面是原始的蛋白。
python check_diff_online.py A152E A484E_rbd.pdb omi_rbd.pdb 152 A484E_check.pdb omi_check.pdb
设定正确的二硫键,分别加载两种水盒,输出protein和complex的拓扑学文件和坐标文件。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
ligand = loadpdb omi_ace2.pdb
bond ligand.308.SG ligand.316.SG
bond ligand.519.SG ligand.536.SG
bond ligand.705.SG ligand.717.SG
m1 = loadpdb omi_check.pdb
bond m1.4.SG m1.29.SG
bond m1.47.SG m1.100.SG
bond m1.59.SG m1.193.SG
bond m1.148.SG m1.156.SG
m2 = loadpdb A484E_check.pdb
bond m2.4.SG m2.29.SG
bond m2.47.SG m2.100.SG
bond m2.59.SG m2.193.SG
bond m2.148.SG m2.156.SG
w1 = loadpdb rbd_wat.pdb
w2 = loadpdb wat1.pdb
protein = combine {m1 m2 w1}
complex = combine {m1 m2 ligand w2}
set default nocenter on
setBox protein vdw {43.215 53.421 59.922}
savepdb protein protein.pdb
saveamberparm protein protein.parm7 protein.rst7
setBox complex vdw {64.171 75.490 114.587}
savepdb complex complex.pdb
saveamberparm complex complex.parm7 complex.rst7
quit
使用parmed处理protein.parm7和complex.parm7,以保证正确的位置。
parmed protein.parm7 <<_EOF loadRestrt protein.rst7 setOverwrite True tiMerge :1-193 :194-386 :152&!@CA,C,O,N,H,HA,CB :345&!@CA,C,O,N,H,HA,CB outparm merged_A484E_protein.parm7 merged_A484E_protein.rst7 quit _EOF parmed complex.parm7 <<_EOF loadRestrt complex.rst7 setOverwrite True tiMerge :1-193 :194-386 :152&!@CA,C,O,N,H,HA,CB :345&!@CA,C,O,N,H,HA,CB outparm merged_A484E_complex.parm7 merged_A484E_complex.rst7 quit _EOF
正确获得这些文件后,使用:
python auto_gene_inp_run.py 152 193 CA,C,O,N,H,HA,CB A484E
来生成tmpl文件(run.tmpl文件需要上传),并将tmpl文件转成真正的ti文件放进文件夹,同时使用slurm提交最小化,加热和运行步骤(5ns)。
审核编辑 黄昊宇
- 相关推荐
- 热点推荐
-
亥姆霍兹自由能判据2023-08-29 6694
-
一种高效的合金设计新策略:耦合计算热力学方法和机器学习技术2022-07-28 3359
-
详细说明热力学中影响热设计的因素2021-11-10 1245
-
基于热力学的流量计的原理及设计2018-09-30 1628
-
什么叫热力学温度_热力学温度与摄氏温度的关系详解2018-06-08 23811
-
浅析MCR框架的Web热力学数据库架构模式及其优势2017-10-23 3839
-
《新热力学第二定律极其系列专利试验、分析科研成果》的链接:发明家方面军2012-01-27 2286
-
超级电容器的划时代应用——新热力学第二定律2012-01-19 2402
-
聚合物溶液热力学模型的评述2010-01-01 1001
-
链状分子流体混合物热力学性质的预测模型2009-05-07 955
-
热力学温度-摄氏温度变送器电路2009-02-23 2556
全部0条评论

快来发表一下你的评论吧 !
