

CO2转化为C2产物的高效光催化剂的设计研究
描述
成果展示
单原子催化剂(SACs)在光催化CO2转化为C2产物方面表现出了巨大的潜力,但是气态多碳烃产物的生成仍具有挑战性。以前,单个原子的载体由多个元素组成,由于单原子位点的配位环境多样化,难以控制,使得C-C偶联困难。
基于此,清华大学李亚栋院士和王定胜副教授、中山大学胡卓锋副教授(共同通讯作者)等人报道了一种由在红磷(RP)负载金(Au)单原子的单原子催化剂(Au1/RP SAC),并用于诱导C-C偶联,其中RP是由单一元素组成的具有均匀结构、较低电负性和更好吸收CO2能力的载体。
Au单原子附近的富电子磷原子可以作为CO2活化的活性位点,使得Au单原子能有效降低C-C耦合的势垒,进而提高形成乙烷(C2H6)的反应动力学。值得注意的是,在没有牺牲剂的情况下,Au1/RP的C2H6选择性和周转频率分别达到96%和7.39 h-1,几乎代表了目前已报道的C2化学合成最好的光催化剂。本研究将为CO2转化为C2产物的高效光催化剂的设计提供新的思路。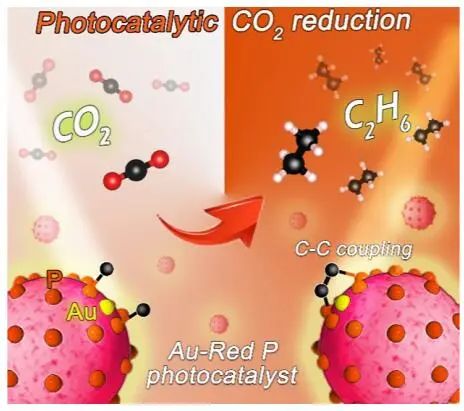
图1. Au1/RP光催化CO2转化为乙烷
背景介绍
目前,利用光催化剂可以高效催化CO2转化为CO、甲烷等C1化合物,但在光催化CO2还原中很难产生双碳(C2)产物,因为其需要C-C偶联,C-C键的形成需要克服比C-H键和C-O键形成更大的反应势垒。
虽然大量报道了在CO2还原过程中制备乙醇和乙酸等液态氧合多碳产物,但很少有关于CO2还原生产气态多碳烃产物,例如在无牺牲剂下生成C2H6。
光催化CO2还原生产C2H6比较困难,因为需要打破非常稳定的C-O键,且选择性通常低于30%。因此,开发有效的光催化剂将CO2转化为气态多碳烃产物是一项非常重要和具有挑战性的任务。
目前,许多工作都集中在改变单金属原子的类型和配位结构上,以实现CO2的高效率转化,但对于金属单原子作为活性位点的催化剂,一个C1中间体需要解吸并迁移到另一个C1中间体上进行C-C偶联,必然降低了C2的生成速率和选择性。
因此,还需要考虑CO2还原中间体与载体之间的相互作用。红磷(RP)是一种单质磷光催化剂,制备简单、稳定性高,在光催化领域具有广泛的应用潜力。
RP具有以下优点:
(1)RP的组成元素单一,可为金属单原子提供相同的配位环境;
(2)磷(P)具有较低的电负性,P元素的配位可以控制金属单原子的电子密度;
(3)富电子的P原子可有效吸附CO2,并通过Lewis酸碱相互作用与CO2中的O相互作用,促进C-O键的裂解,生成较大的C1中间体。
因此,设计和构建RP负载金属单原子以促进C1中间体的电荷分离和偶联是实现高活性和高选择性制备C2产物的潜在途径之一。
图文解读
合成与表征
作者首先合成了块状多孔RP,然后在搅拌下将RP分散在HAuCl4溶液中,搅拌半小时。接着,通过冷冻干燥过程进行染色。
最后,在H2气氛中煅烧RP以获得Au1/RP SAC。TEM图像显示,Au1/RP具有多孔形态,未观察到Au纳米颗粒的存在。
同时,Au1/RP的HAADF-STEM图像显示未发现金属颗粒。P和Au元素均匀分布在收集区域,Au原子均匀分布在RP载体上。
图2. Au1/RP催化剂的形貌表征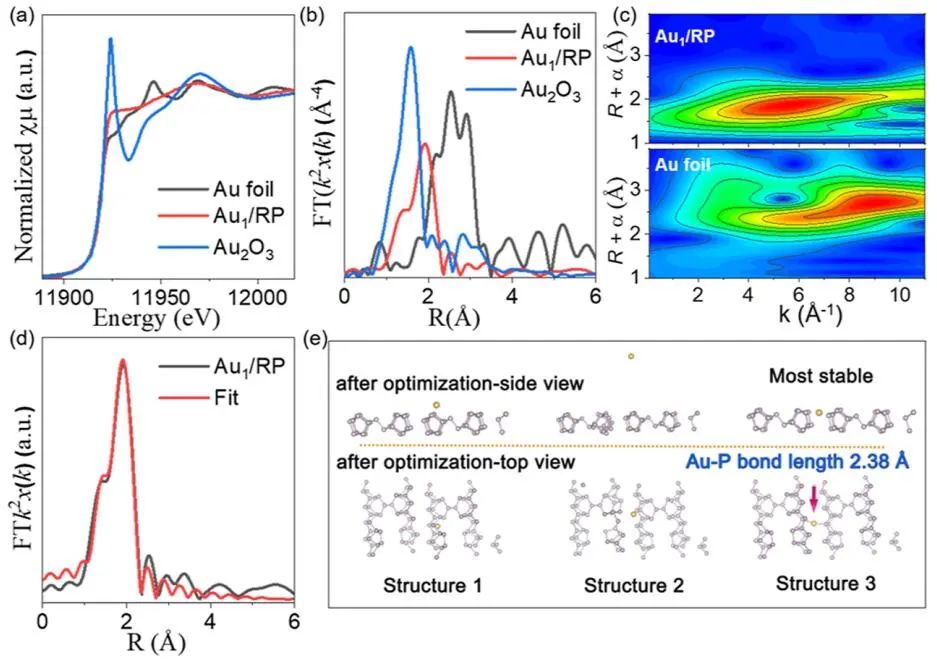
图3. Au1/RP催化剂的结构表征
CO2还原性能
在Au1/RP上,CO和CH4的产率分别仅为0.02和0.03 μmol g-1 h-1,而C2H6的产率高达1.32 μmol g-1 h-1,同时选择性高达96%,其产率和选择性优于大多数最先进的光催化剂,表明Au1/RP对C2H6的生成具有良好的选择性。此外,在RP上负载了Pt、Ir、Pd和Au-Pd不同的金属单原子,只有Au对C2H6的产生表现出较高的活性,证实Au的作用是生成C2H6。
此外,Au1/RP制取C2H6的周转频率(TOF)可达到0.052 h-1,高于大多数光催化剂。如果将CO2的吸附量作为活性位点,计算出Au/RP的TOF可达7.39 h-1的极高值。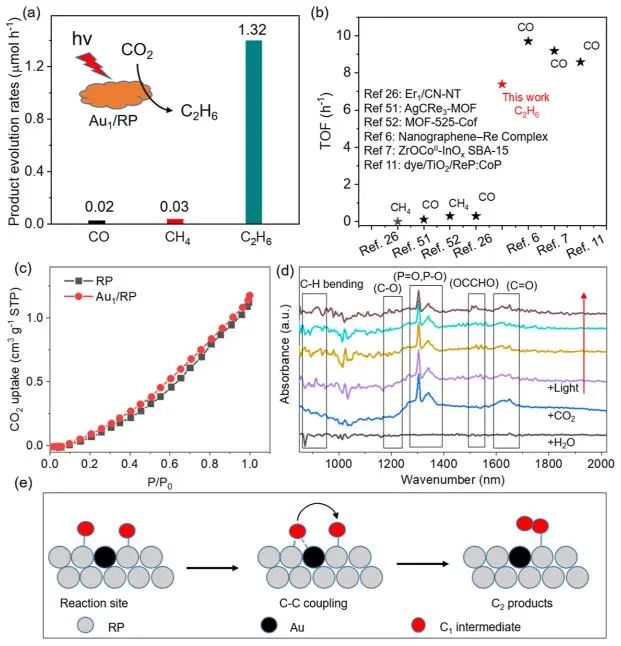
图4. 评估CO2还原性能
研究C-C偶联起源
在文中,作者构造了两个C分离连接的态,研究了六个中间态。在计算中有8个状态,能量变化图如下:刚开始(状态1到状态4)能量迅速下降,两个C原子越来越近(状态1到状态4),然后从状态4到状态5,能量保持在同一水平。在此期间,形成了C-C键,两个C原子也与附近的P原子成键。
随后,一个C原子离开P原子,变得更接近Au原子(状态6到状态8),可能是由于Au原子对C原子的吸引。在此期间,能量再次下降。总之,C-C偶联是有利的,表明Au1/RP有利于C2H6的形成。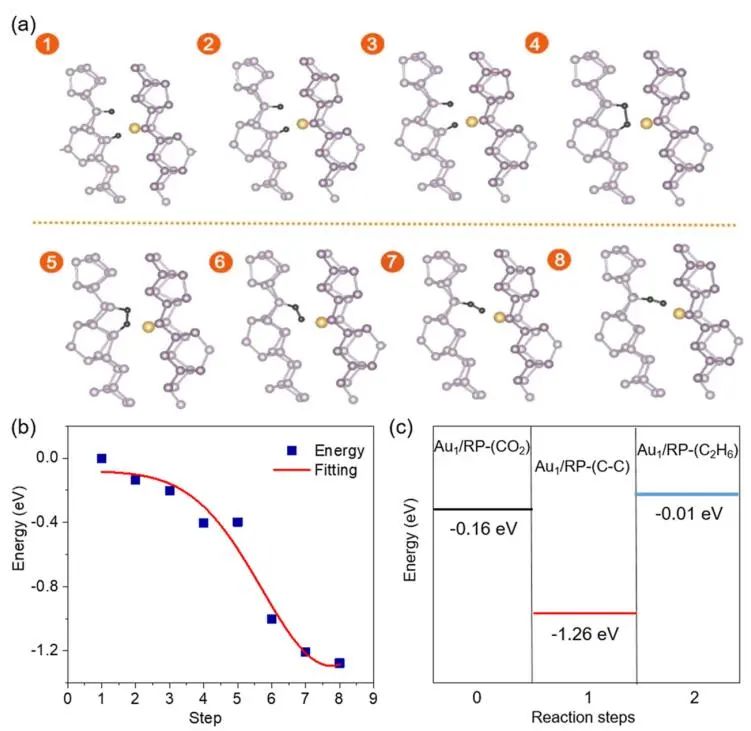
图5. C-C偶联起源的研究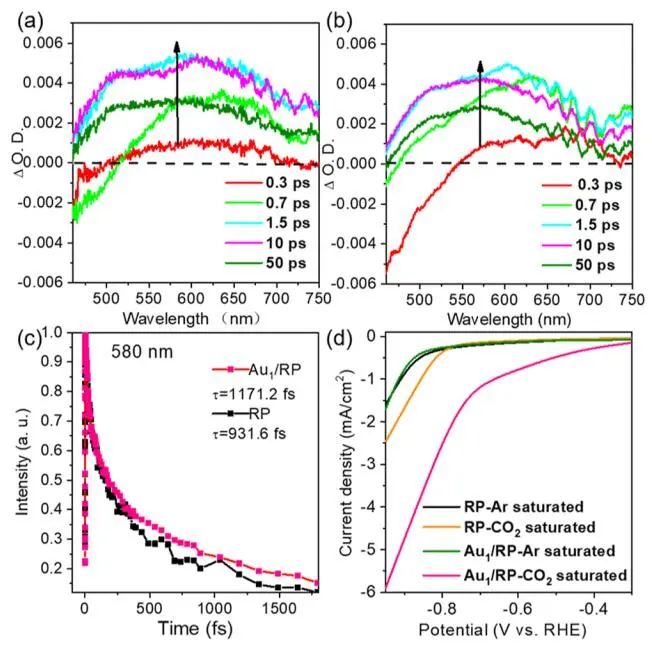
图6. 载流子动力学
审核编辑:刘清
-
反向电子转移!双-单原子催化剂助力CO2光还原2023-08-29 3234
-
可回收光催化剂的结构调控与催化性能研究2023-08-09 1964
-
大面积二维Cu2Te垂直阵列催化剂助力CO2电还原2023-07-17 2575
-
CO2辅助生成富含晶界的Cu催化剂实现高效CO-CO偶联2023-03-17 2116
-
金属簇催化剂的CO2转化反应性和循环性2023-01-09 1662
-
揭示卤素掺杂Sn基催化剂促进CO2电还原制甲酸盐原因2022-12-29 4237
-
CdS/ZnS 1D/2D异质结光催化剂用于高效光催化制氢2022-12-20 3286
-
基于氨基的D-A结构助力CO2转化为CH42022-12-09 2652
-
红磷负载Au单原子实现CO2光还原为C2H62022-12-05 2972
-
构建TiO2/FePS3梯形异质结促进高效光催化析氢2022-10-17 4797
-
蜂窝状多孔结晶异质电催化剂实现高效的CO2吸附/活化2022-09-30 4294
-
一种用于低浓度CO2的有效光还原的技术2022-09-02 2981
-
生成CO的催化剂与Cu之间的相互作用2022-08-22 4052
-
碱性醇类燃料电池新型催化剂的研究2011-03-11 1987
全部0条评论

快来发表一下你的评论吧 !
