

红磷负载Au单原子实现CO2光还原为C2H6
描述
研究背景
光催化CO2转化因其在高附加值化学品生产中的潜在应用而受到广泛关注。在光催化CO2还原中,生产双碳 (C2) 产品非常困难,C-C键的形成需要克服比C-H键和C-O键形成更大的反应能垒。在没有牺牲剂的条件下,通过CO2还原生产气态多碳烃类产物(如C2H6)的报道很少。
具有独特电子结构和丰富活性位点的单原子光催化剂引起了大量的关注。金属单原子活性位点的设计和构建可以加速光生电荷的转移和反应中间体的偶联。然而,对于以金属单原子为活性位点的催化剂,一个C1中间体需要脱附并迁移到另一个C1中间体并进行C-C偶联,这不可避免地降低了C2的生成速率和选择性。
因此,如果活性位点也涉及单个金属原子附近的载体,将会更加有利于反应的进行。因此,还需要考虑CO2还原中间体与载体之间的相互作用。载体的类型也极大地影响了C-C键的耦合。具有丰富活性位点的合适载体可以有效缩短光生电子和C1中间体的迁移距离,促进C2产物的生成。
然而,迄今为止,常见的半导体催化剂载体由多种元素组成。这些元素作为单原子配位元素,使得单原子位点的配位环境多样化且难以控制。相比而言,结构确定的单元素载体在催化机理研究中具有巨大的优势。
红磷(RP)是一种磷单质光催化剂,由于其制备简单、稳定性高等优点,在光催化领域具有广阔的应用潜力。RP具有下列优点: (i) RP的组成元素单一,可以为金属单原子催化剂提供统一的配位环境。
(ii)与常见的单原子配位元素(N、O、S等)相比,磷(P)的电负性较低,因此可以通过P元素的配位来控制金属单原子的电子密度。(iii)富电子的P原子可以有效地吸附CO2,并通过路易斯酸碱相互作用,与CO2中的O相互作用,促进C-O键断裂,产生C1中间体。因此,设计构建RP负载的金属单原子催化剂,以促进C1中间体的电荷分离和偶联,是实现高活性和高选择性地制备C2产物的潜在途径之一。
成果简介
清华大学李亚栋院士、王定胜副教授和中山大学胡卓锋副教授(共同通讯作者)通过在红磷 (Au1/RP)上引入一个Au单原子来诱导C-C偶联。P具有较低的电负性,可以更好的吸附CO2。Au单原子附近的富电子P原子可以作为吸附CO2并促进CO2裂解的活性位点。Au单原子可以有效降低C-C偶联的能垒,促进C2H6的形成。
研究亮点
1. Au1/RP催化CO2光还原生成C2H6的选择性和转换频率分别达到96%和7.39 h –1 ;
2. Au单原子不仅可以促进光生电荷的转移,也可以诱导C1中间体偶联形成C2产物。
图文导读
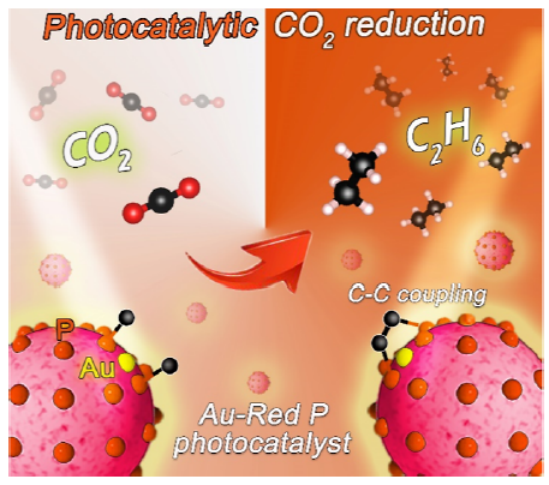
图 1. Au 单原子与红磷形成的界面辅助催化C-C偶联。
将Au单原子负载到RP催化剂上,在没有牺牲剂的情况下,光催化CO2还原为乙烷(图1)。

图 2. (a) Au1/RP的TEM。Au1/RP的AC HAADF-STEM (b) 和相应的放大图像 (c) 。(d) Au1/RP的 HAADF-STEM 图像。比例尺为100 nm。(e) Au1/RP的Au和P的元素映射图。
如图2a所示,Au1/RP的TEM表明,Au1/RP具有多孔形貌,未观察到Au纳米颗粒的存在。如图2 b、c所示,发现许多孤立的亮点,这些亮点被识别为Au 单原子。Au1/RP的HAADF-STEM显示,没有发现金属颗粒(图2d)。如图2e所示, Au原子均匀分布在RP骨架上。
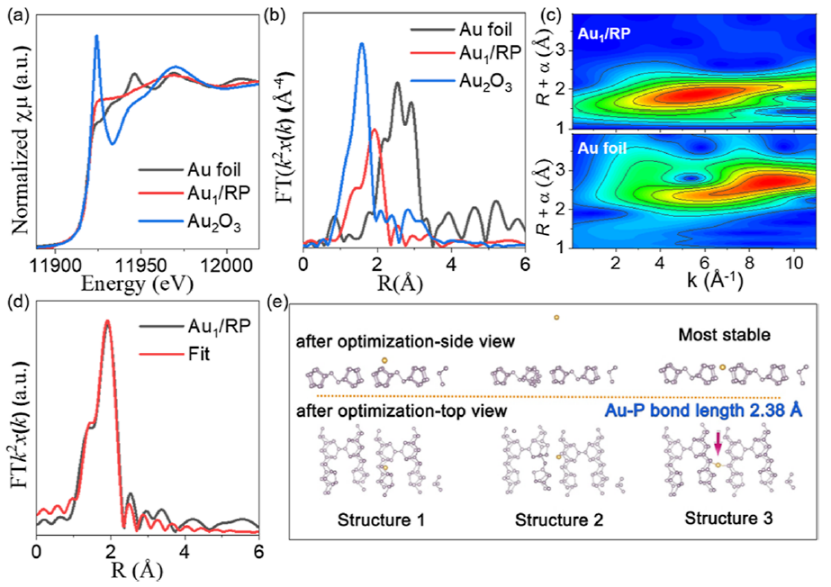
图 3. (a) Au K边XANES光谱;(b)傅里叶变换图;(c)小波变换EXAFS 光谱;(d) Au1/RP在R空间的EXAFS拟合曲线;(e) Au1/RP三种可能的结构。
如图3a所示, Au1/RP的XANES光谱吸收强度介于Au箔和Au2O3之间,表明Au1/RP催化剂中的Au物种带正电,价态为在0和+3之间。从EXAFS分析(图3b)可以看出,Au1/RP在1.83 Å处显示出突出的峰。由于Au-Au 配位 (>2.5 Å) 的位置没有观察到峰,这表明Au1/RP 中不存在 Au 粒子。
从Au箔(图3c)可以分析出 R 空间中2.7和1 Å处的最高强度峰对应于Au-Au和Au-O配位。然而,Au1/RP仅检测到R空间中 1.8 Å处的峰值强度最大值(图3c),这归属于Au-P配位。如图 3d所示,Au1/RP的Au单原子配位数约为2。Au单原子与两个P原子配位形成Au-P2结构体,平均键长约为 2.34 Å。
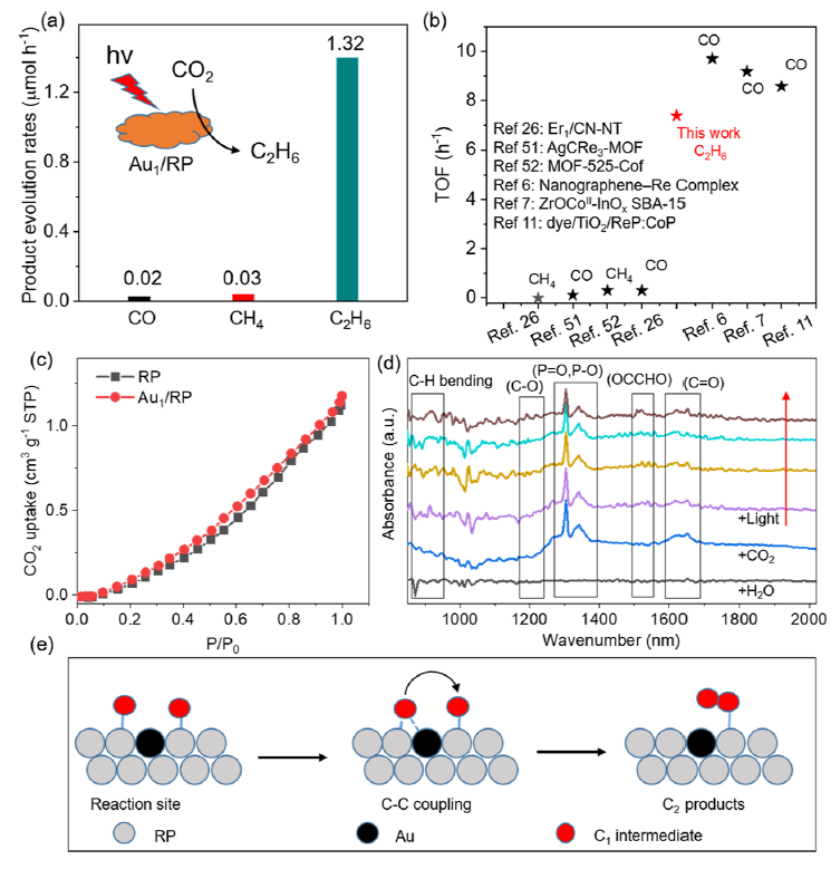
图 4.(a) Au1/RP 在CO2还原反应中的光催化活性。(b) Au1/RP光催化CO2还原的TOF与近期报道的比较。(c) 室温下,RP和Au1/RP的CO2吸附。(d)用于CO2还原反应的Au1/RP的原位DRIFTS光谱。(e) Au1/RP催化C-C偶联的反应机理。
CO和CH4在Au1/RP上的产率分别仅为 0.02 和 0.03 μmol g –1 h –1,而 C2H6的产率高达 1.32 μmol g –1 h –1(图 4a),选择性高达 96%。Au/RP的 TOF可达到7.39 h –1(图 4 b)。如图4c所示,负载Au单原子后,样品对CO2的吸附能力没有提高,证明Au不是CO2的吸附位点。如图4d所示,在800–1700 cm –1处观察到一些峰。1520 cm –1处的峰可归因于OCCHO伸缩振动。900 cm –1处的峰与C–H的振动有关。
当添加CO2时,可以观察到1357 和 1621 cm –1处的峰。这些峰分别对应于 P=O/P-O 和 C=O 的振动。这证明RP能有效吸附CO2分子,是CO2的活化位点。因此,可以推测Au1/RP催化C-C偶联的反应机理。如图 4 e所示,Au单原子促进C1中间体在P反应位点发生解吸和迁移,然后在另一个P 位点进行 C-C 偶联。
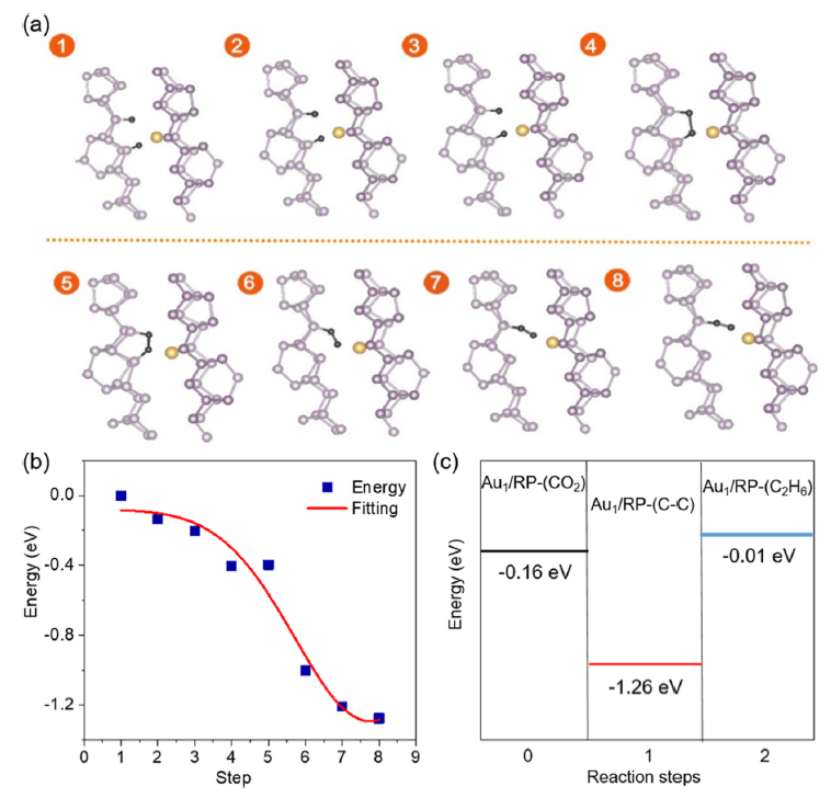
图 5. 吸附了C-C中间体的Au1/RP的 (a) 结构变化;(b) 能量变化。(c) 将CO2光催化还原为C2H6的三个重要步骤的能量变化。
光催化CO2还原反应过程中的能量变化如图5a,b所示。 在该反应的初始阶段,反应体系的能量迅速下降,相邻C原子间距变小(状态1到状态4)。从状态4到状态5,能量保持在同一水平。在此期间,形成了C-C键,两个C原子也与附近的P原子成键。
随后,一个C原子离开P原子并靠近Au原子(状态 6 到状态 8),这可能是由于Au对C原子的吸引。在此期间,能量再次下降。因此,Au1/RP界面有利于C-C原子偶联及C2H6形成。作者发现,反应体系的能量从初始状态 (-0.16 eV) 下降到稳定的中间状态 (-1.26 eV),表明CO2偶联很容易发生在 Au1/RP界面(图 5c)。
然而,从中间态到最终态,体系能量增加,表明该反应需要克服能垒才能形成C2H6。因此,理论计算可以证实,单个金属原子可以通过调控活性位点的电子结构,促进活性位点上C1中间体的解吸和迁移,进而与另一位点的C1中间体偶联,导致较低的C-C偶联能垒。
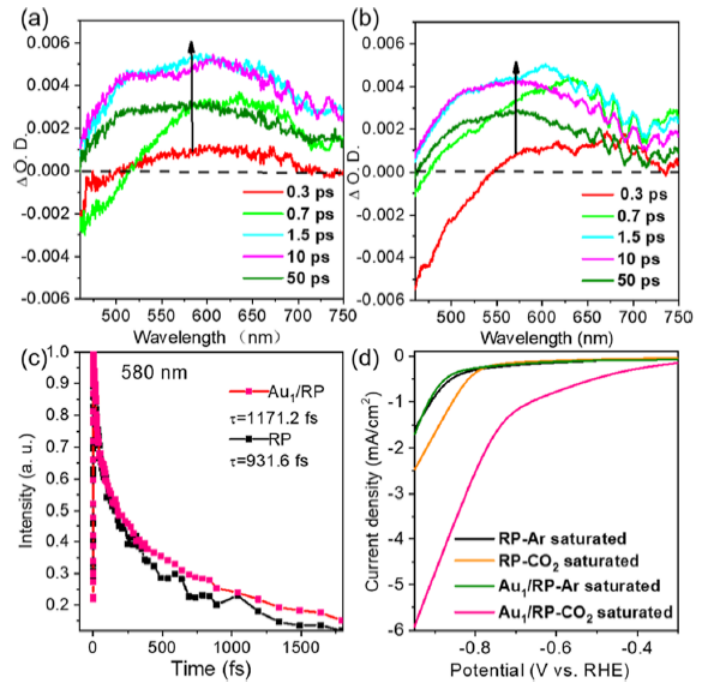
图 6. (a) RP和 (b) Au1/RP的瞬态吸收光谱。(c) RP和 (b) Au1/RP在580 nm波长处的归一化衰变曲线。(d) RP和Au1/RP在Ar或CO2饱和的KHCO3电解质中的线性扫描伏安曲线。
RP的瞬态吸收光谱如图6a所示。在开始时(0.3ps),可以观察到明显的正吸收峰,这对应于发射态的光激发电子。随着时间的推移(0.3-1.5 ps),这个峰大大增加,表明光生电子的积累。随后,在50 ps时,由于空穴和电子的复合,观察到轻微的衰减。
对于Au1/RP,正吸收峰强度的增加甚至更快(图 6 b)。在0.7 ps时达到最高,一直保持到10ps。这表明Au单原子促进了光激发电子的产生。此外,光激发电子的寿命也可以通过 580 nm 波长下的归一化单衰变曲线来确认(图6c)。初始上升阶段表示电荷载流子从基态到激发态是瞬时产生和直接激发的。
随后的下降阶段是由于电子和空穴的复合。Au1/RP比RP衰减得更慢,对应于更慢的电荷复合率。Au1/RP中的电子寿命为1171.2 fs,而RP中的电子寿命为931.6 fs。如图6d所示, RP和Au1/RP 有类似的曲线,由于饱和Ar中的阴极电流主要来自析氢反应,该结果表明Au不会促进析氢反应。相比之下,在CO2饱和的电解质中,Au1/RP的电流密度增加明显更快,并且起始电位也更早。
总结与展望
作者用RP负载Au单原子,将CO2有效的光催化还原为C2H6。通过各种表征技术,包括AC HAADF-STEM、XAFS光谱和理论计算,分析了Au单原子的结构和化学性质。实验和理论计算结果证实,RP上的Au原子不仅能有效促进C-C偶联,还能促进光生电荷的转移,从而促进CO2还原为C2H6。该研究为C1转化提供了一种在原子水平上设计催化剂的活性位点,从而实现C-C偶联的有效方法,对基于太阳能的燃料制备具有重要意义。
文献链接
Ou Honghui et.al Atomically Dispersed Au-Assisted C–C Coupling on Red Phosphorus for CO2 Photoreduction to C2H6 (J. Am. Chem. Soc.2022, DOI:10.1021/jacs.2c09424)
文献链接:
https://pubs.acs.org/doi/full/10.1021/jacs.2c09424
审核编辑:刘清
-
电化学CO2甲烷化的进展与挑战2023-09-25 4223
-
反向电子转移!双-单原子催化剂助力CO2光还原2023-08-29 3288
-
基于STM32单片机的CO2检测系统设计2023-07-19 5604
-
CO2辅助生成富含晶界的Cu催化剂实现高效CO-CO偶联2023-03-17 2168
-
分子催化剂助力酸性条件下的CO2还原2023-01-10 3172
-
如何更好地报道CO2电还原的性能?2023-01-08 2929
-
在熔盐中利用液态金属锡阴极实现整体碳中和电化学还原CO22022-12-30 3435
-
揭示卤素掺杂Sn基催化剂促进CO2电还原制甲酸盐原因2022-12-29 4320
-
黄洪伟AM:P调制氮化碳中的Cu-N4位点实现CO2光还原为C2H42022-12-01 3041
-
CO2转化为C2产物的高效光催化剂的设计研究2022-11-25 4709
-
CO2电解的安培级电流密度对于实现多碳燃料的工业生产2022-10-18 3603
-
一种用于低浓度CO2的有效光还原的技术2022-09-02 3052
-
利用STM32的USART2串口采集CO2传感器数据2022-02-22 1974
-
稳态法联合检测CO2和O2的全固态电化学传感器研究2009-06-23 892
全部0条评论

快来发表一下你的评论吧 !
