

双原子Fe-Mo位点超强酸性ORR活性
描述
01
导读
众所周知,阴极氧还原反应(ORR)涉及多电子转移步骤,使得反应动力学十分缓慢,进一步限制了质子交换膜燃料电池等清洁能源转化装置的输出功率。目前,Pt仍然是广泛应用的商业电催化剂之一,但因其使用成本较高、储量稀缺而无法满足日益增长的商业需求。Fe-N-C催化剂因其具有高的本征ORR活性,而被认为是最具有应用前景的非贵金属催化剂之一。然而,在酸性介质下,Fe-N-C催化剂的ORR活性以及耐久性仍然不能达到实用指标。
另一方面,近年来,双原子催化剂的发展如火如荼。以Fe-N-C催化剂为基础,近年来已逐渐开发出了Fe-Mn、Fe-Co、Fe-Ni等双原子催化剂,在相关的电催化体系也显示出了优于单原子催化剂的电化学性能。与后过渡金属相比,将前过渡金属引入Fe-N-C的研究报道相对较少。前过渡金属具有的更多的未占据d轨道以及亲氧特性,有望地进一步调节Fe中心的ORR活性。
02
成果背景
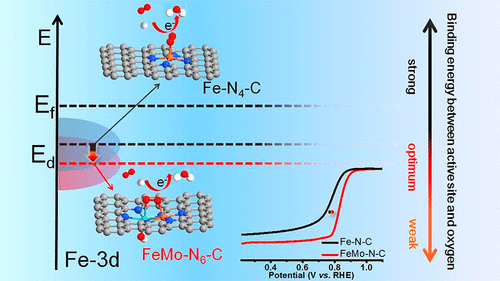
首都师范大学孙文明,清华大学王定胜、李亚栋院士等人成功在N掺杂碳基底上引入了Fe-Mo双原子位点,并记为FeMo-N-C。研究发现,引入的Fe-Mo原子对可以有效地调控Fe的电子构型,并使Fe的d带中心发生负移,从而削弱对ORR中间体的吸附,最终促进ORR。
因此,FeMo-N-C在酸性电解质中表现出良好的ORR活性,起始电位高达0.98 V,半波电位达0.84 V。相关成果以《Regulating the FeN4 Moiety by Constructing Fe–Mo Dual-Metal Atom Sites for Efficient Electrochemical Oxygen Reduction》为题发表于国际顶尖杂志《Nano Letters》上。
03 关键创新
(1)本文首次设计并制备了一种新型的Fe-Mo双原子催化剂,经过详细的结构表征,证实了Fe、Mo主要以FeMo-N6的结构形式高度分散于碳载体上;
(2)在酸性介质下,FeMo-N-C表现出优异的ORR性能,其半波电位达到了0.84 V,远优于Fe-N-C以及其他文献所报道的非贵金属催化剂的性能;
(3)理论计算揭示了在新型的FeMo-N6位点中, 引入的Mo可以调节Fe中心的电子构型,使d带中心负移,从而削弱了对ORR中间体的吸附,加速了ORR。
04
核心内容解读
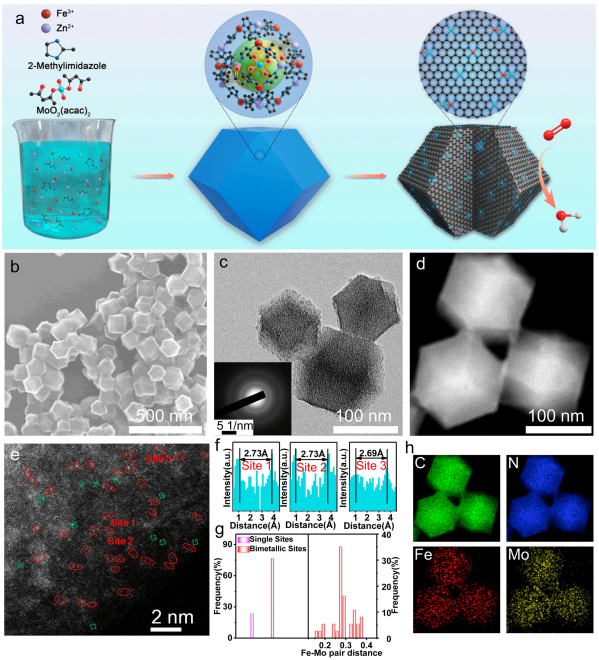
图1 合成示意图及结构表征:(a) FeMo-N-C的合成示意图;(b) FeMo-N-C的SEM图像;(c) TEM图像和相应的选取电子衍射图案;(d) HAADF-STEM图像;(e)像差校正HAADF-STEM图像;(f,g)对图e的选定区域进行原子强度分析以及Fe-Mo双位点的占比、原子间距进行统计;(h)图d相应的EDS映射。
本文采用主客体策略合成了含有Fe-Mo双原子位点的FeMo-N-C催化剂。如图1a所示,乙酰丙酮钼被封装在原位生长的FeZn-ZIF骨架中,将所得的Mo-FeZn-ZIF进行热解可得多孔碳基催化剂。图1b、c的SEM、TEM图像显示,FeMo-N-C仍然保留了十二面体形貌,直径约100 nm。
相应的选区电子衍射(SAED)图案表明该多孔碳的结晶度相对较高,有利于增强导电性。图1d的HAADF-STEM图像显示该多孔碳上未存在明显的纳米颗粒。采用像差校正的HAADF-STEM图像对金属原子的分散形式进行辨别,如图1e所示,可以看到碳基底上分布大量的Fe/Mo双原子对(红圈标识),其对应的电子能量损失谱(EELS)表明这些原子对由Fe和Mo所构成。
另一方面,少量单个Fe或Mo原子用绿圈标出。对图1e中标记的区域1、2和3的Fe-Mo双金属位点距离进行测量,如图1f所示,其平均间距约为0.27 nm。图1g的统计结果显示,大部分(~70%)亮点的原子间距为0.3 nm,也说明了催化剂中具有大量的Fe-Mo双原子位点。通过相应的EDS映射(图1h),进一步验证了Fe、Mo和N元素在碳载体中均匀分布。
最后,ICP-MS测试结果显示Fe和Mo的负载量分别为0.78和0.71 wt %。
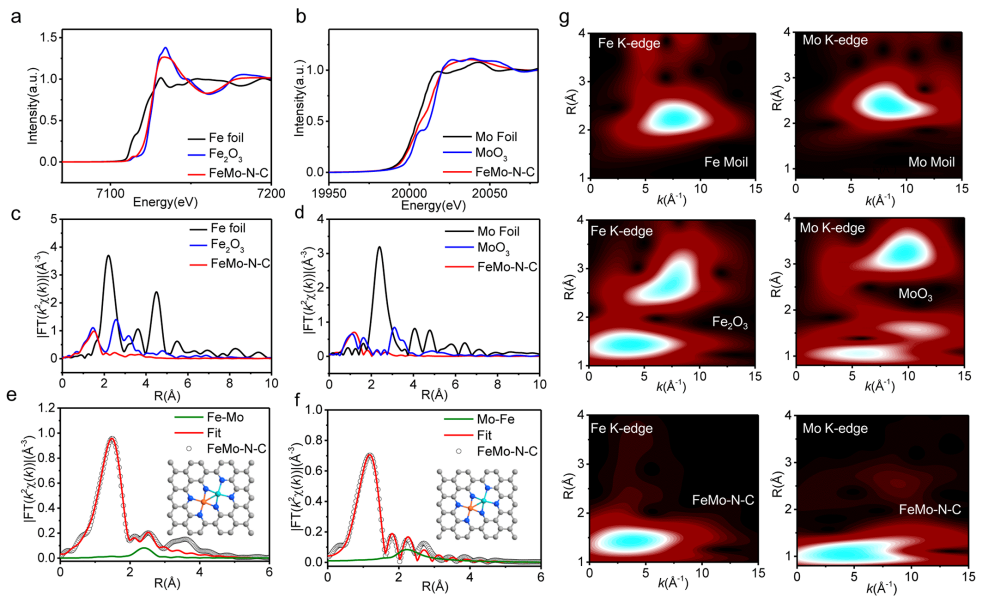
图2 基于XAFS的结构分析:(a) Fe的K边XANES谱图;(b) Mo的K边XANES谱图;(c,e) Fe的K边FT-EXAFS谱图及其拟合结果;(d,f) Mo的K边FT-EXAFS谱图及其拟合结果;(g)小波变换。
采用XAFS对FeMo-N-C催化剂的原子精细结构进行解析。如图2a的Fe的K边XANES谱图所示,FeMo-N-C的近边吸收谱线介于Fe与Fe2O3之间,说明其Fe物种处于正价态(+0<δ<+3)。类似地,在Mo的K边XANES谱图中,FeMo-N-C的近边吸收谱线介于Mo与MoO3之间,即Mo物种处于正价态(+0<δ<+6)。
图2c、d的Fe、Mo的K边FT-EXAFS谱图显示,FeMo-N-C分别在~1.5 Å、1.2 Å处出现主峰,分别对应Fe-N、Mo-N配位,同时均在2.2 Å、2.4 Å处未发现明显的Fe-Fe、Mo-Mo配位峰,从而支持了电镜观察结果。对谱图进行拟合,结果如图2e、f所示,结果显示说明Fe-N、Mo-N的配位数均为4,且谱图均在~2.7 Å处出现了弱配位峰,结合上述统计结果,可以将其归为Fe-Mo配位峰。
图2g的小波变换显示,FeMo-N-C中未发现Fe-Fe与Mo-Mo信号,进一步支持了这一结果。因此,Fe、Mo以FeMo-N6的结构形式分散于碳载体上。
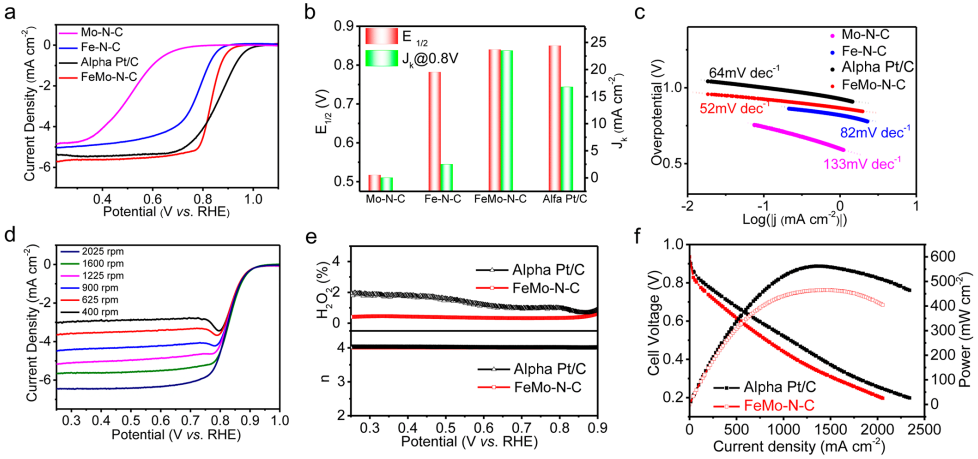
图3 电催化ORR性能:(a)在O2饱和的0.1 M HClO4溶液下的LSV曲线;(b)半波电位以及在0.8 V下的动力学电流密度比较;(c)不同催化剂的Tafel斜率比较;(d) FeMo-N-C在不同转速下的极化曲线;(e)基于RRDE测试得到的转移电子数与H2O2产量;(f)基于FeMo-N-C的H2-O2燃料电池的放电极化曲线与放电功率密度。
采用标准的三电极测试体系研究了催化剂的ORR性能。在含饱和O2的0.1 M HClO4溶液中,图3a显示了各催化剂的极化曲线。其中,FeMo-N-C显示出了优异的ORR性能,其起始电位高达0.98 V,同时半波电位达0.84 V,优于Fe-N-C、Mo-N-C,同时也是目前报道的最佳的非贵金属基酸性ORR电催化剂之一。
同时,如图3b所示,在0.8 V下FeMo-N-C的动力学电流密度为23.5 mA cm-2,远大于Fe-N-C和Mo-N-C的动力学电流密度之和,说明Fe-N-C和Mo-N-C位点之间存在协同作用。由极化曲线衍生的Tafel曲线(图3c)显示,FeMo-N-C表现出低至52 mV dec-1的Tafel斜率,具有最快的ORR动力学。
对不同转速下的极化曲线进行K-L方程拟合,得到FeMo-N-C的转移电子数为3.99。同时,图3e的RRDE测试显示n接近4,H2O2产率低于1.0%。两种测试共同说明了FeMo-N-C遵循了高效的四电子反应机制。
最后,将FeMo-N-C作为阴极催化剂,并用于H2-O2燃料电池中,如图3f所示,该电池的峰值功率密度为460 mW cm-2,约为Pt/C基燃料电池的83%,并超过了大多数非贵金属催化剂所报道的输出性能。
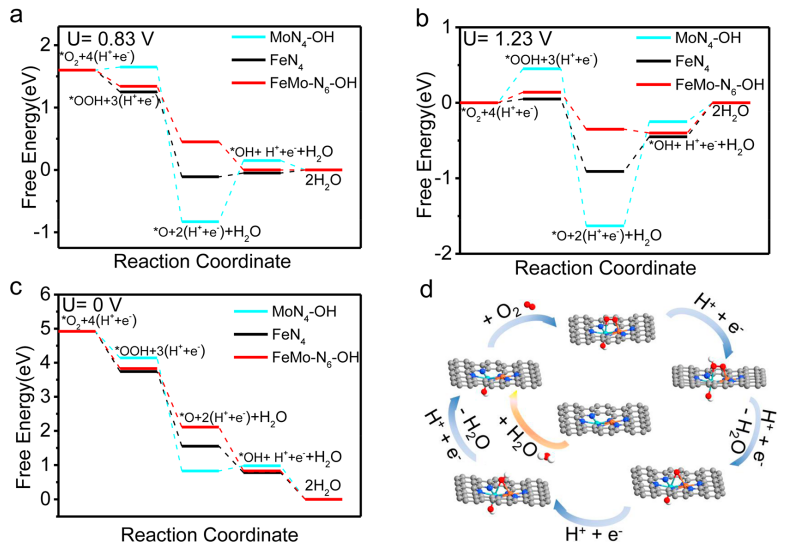
图4 催化机理分析:(a-c)在不同外加电位下,FeMo-N-C、Fe-N-C与Mo-N-C的ORR自由能分布;(d) FeMo-N-C的ORR机制。
采用DFT计算研究了FeMo-N-C的ORR机制。首先,在整个工作电位下,*OH在FeMo-N-C上的吸附是热力学自发的,因此形成了FeMo-N6-(OH)构型。类似的结果也存在于MoN4位点上。相比之下,FeN4位点上不存在轴向OH配体吸附。如图4a所示,在U=0.83 V,FeMo-N6-(OH)位点在整个ORR过程中均是热力学有利的,而MoN4-(OH)位点与FeN4位点在*O向*OH的转化步骤均为热力学吸热过程。
另外,根据图4b、c的计算结果,可以看出在整个工作电位下,MoN4-(OH)位点上均存在吸热步骤,导致其ORR活性较差;而FeN4位点虽然表现出更有利的O2质子化过程,但对于后续反应中间体的吸附过强,导致其ORR活性低于FeMo-N6-(OH)位点。因此,理论计算分析验证了这种新型的FeMo-N6-(OH)位点的优势,从而极大地促进了ORR性能,其反应机制如图4d所示。
05
成果启示
本文设计了一种新型的双原子Fe-Mo电催化剂,并在酸性电解质下表现出超强的ORR活性。由于O2更倾向于以桥键形式吸附于Fe-Mo原子对上,O-O键更容易发生断裂,从而有利于促进ORR。另一方面,引入的Mo可以调节Fe中心的电子构型,使d带中心负移,从而削弱了对ORR中间体的吸附,加速了ORR。因此,FeMo-N-C在半电池与全电池的测试过程均显示出了优异的性能。总之,该工作为双原子催化剂的设计提供了新的见解,加速了非贵金属催化剂在燃料电池的应用。
审核编辑:刘清
-
ORR逻辑或操作指令相关资料分享2021-12-22 1707
-
水基磁流体的制备及Fe-13Cr-2.5Mo阻尼合金磁畴的观2009-05-16 1124
-
数据处理指令之ORR逻辑或指令2017-10-18 2301
-
高活性生物质碳负载Fe/Pt单原子双功能催化剂开发2021-02-12 3591
-
一款强酸性电解槽将碳的利用率提高到50%2021-06-17 2164
-
盐模板辅助制备多种过渡金属单原子催化剂用于高效氧还原2022-08-11 2340
-
Mo配位FeCoNiMo碳负载高熵电催化析氧催化剂图文解析2022-09-20 3477
-
平衡配体效应和应变效应提升Pt合金的ORR活性2022-11-07 3604
-
中空碳纳米纤维的双面原子级工程策略构筑“Janus”活性位点助力柔性锌-空气电池2022-11-17 2452
-
“纳米岛”型催化剂突破传统催化剂活性和稳定性的矛盾2022-11-18 1749
-
多掺杂调控局域电荷重排提高ORR/OER双功能催化活性2022-11-21 5163
-
由HPC边缘锚定的单原子Co−N4位点电催化的2e- ORR具有更高选择性2022-11-28 4196
-
原子级分散钌氧化物实现晶格氧参与高效稳定酸性氧析出2023-04-11 6611
-
Adv Mater综述:非贵金属单原子/双原子在光合成的应用2023-05-15 2105
全部0条评论

快来发表一下你的评论吧 !
