

单层NiFeB氢氧化物纳米片中的Ni氧化态转变助力高效OER
描述
研究背景
高氧化态的过渡金属位点(如NiOOH中氧化态大于+3的Ni位点),被认为是析氧反应(OER)的活性位点。通过形成高氧化态的过渡金属位点,可以降低电催化反应的起始电位,从而加快OER的动力学。迄今为止,人们致力于开发高效的镍基催化剂。
然而,通过人为提高Ni的氧化态来提高OER活性,相关研究却相当有限。当镍基催化剂与等离子体纳米颗粒偶联时,在可见光照射下,纳米颗粒中产生了带正电荷的“空穴”,这很容易促进附近的Ni2+氧化成高氧化态的物质,获得更高的OER活性。
Ni基氧化物催化剂与其他金属氧化物(如MoO2和WOx)和非金属(如磷酸盐)偶联,也能有效形成OER所需的高氧化态活性位点。因此,使用这些催化剂可以大幅度提高OER活性。尽管在先前的报道中已取得不错的成果,但OER活性仍远未达到最佳状态。进一步探索促进Ni氧化的有效策略,将OER活性提高至前所未有的水平,仍然是非常迫切的需求。
研究表明,在传统的NiFe氢氧化物催化剂中引入缺电子硼(B),可以有效地促进Ni的氧化态转变,显著提高OER活性。通常,直接从Ni2+中提取电子以形成所需的Ni3+δ是一个困难的过程,只能在高电位下实现。预计在Ni附近的B可能作为参与氧化过程的电子流中转站,参与到Ni氧化过程中。由于其固有的缺电子特性, 转运B位可能作为一个“电子穴”,促进电子从Ni2+位流动,从而允许在较低的电位下形成活性Ni3+δ,从而提高OER活性。
成果介绍
为了验证这一想法,西安交通大学高传博教授和苏州大学程涛教授通过原位水解NiFeB合金纳米粒子合成了单层NiFeB纳米片。拉曼光谱、X射线吸收光谱和电化学测量结果表明, NiFeB氢氧化物纳米片中Ni2+(OH)2转化为Ni3+δOOH的电位低于无B的 NiFe氢氧化物纳米片。
密度泛函理论(DFT)计算表明,Ni原子在加入B后呈现较高的氧化态,表明Ni和B组分之间存在电子相互作用。结果表明,NiFeB氢氧化物纳米片的OER活性达到100 mA cm−2,过电位为252 mV,优于无B的NiFe氢氧化物纳米片的OER活性(过电位为337 mV)和目前报道的大多数镍基催化剂。
虽然硼酸盐已被用作电解质添加剂,过渡金属硼化物已被直接用作OER的催化剂,但B的作用尚不清楚。这项工作揭示了B的缺电子特性在促进Ni氧化态转变方面的作用,为设计先进的金属氢氧化物(或氧化物)催化剂提供了新的机会,提高了电化学裂解水在应用上的可行性。
图文介绍
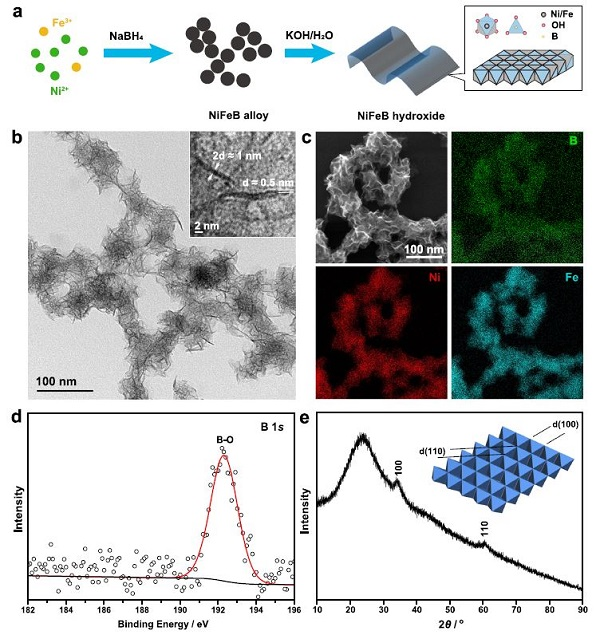
图1. 单层NiFeB氢氧化物纳米片的合成与表征。(a)单层NiFeB氢氧化物纳米片的合成方法。(b)单层NiFeB氢氧化物纳米片的TEM图像。(c)Ni, Fe, B的EDS元素图。(d) B 1s XPS。(e)单层NiFeB氢氧化物纳米片的XRD。
在环境条件下,通过NiFeB合金纳米粒子原位水解,合成单层NiFeB氢氧化物纳米片(图1a)。首先,在超声作用下,用NaBH4在乙醇溶液中还原Ni(NO3)2和Fe(NO3)3,合成了NiFeB合金纳米颗粒。NaBH4既是还原剂又是硼源。接下来,将NiFeB合金纳米颗粒分散在KOH溶液中,将其转化为氢氧化物。
NiFeB合金纳米颗粒经过快速水解,演变为超薄纳米片(图1b)。这些纳米片被缠绕成大量褶皱的束,测量到的皱纹厚度约为1 nm,相当于2层纳米片(图1b,附图)。因此,单层纳米片的厚度约为0.5 nm。能量色散X射线能谱(EDS)元素映射图显示,Ni、Fe和B在纳米片中均匀分布,形成均匀的固溶体,而不是分离相(图1c)。
XPS显示,水解后零价产物消失,仅显示氧化产物(Ni2+、Fe3+和B-O)的信号;这表明纳米片是由NiFeB氢氧化物组成的,即通过桥接O或OH连接的阳离子Ni、Fe、B(图1d)。XRD显示了位于~34°和60°的衍射峰,分别来自金属氢氧化物中MO6(M = Ni, Fe)八面体的(100)和(110)晶面,与NiFe-LDHs中观察到的结构相似(图1e)。
没有观察到其他衍射峰,特别是来自共边MO6八面体的反射峰,这表明纳米片是由单一的MO6层组成的。
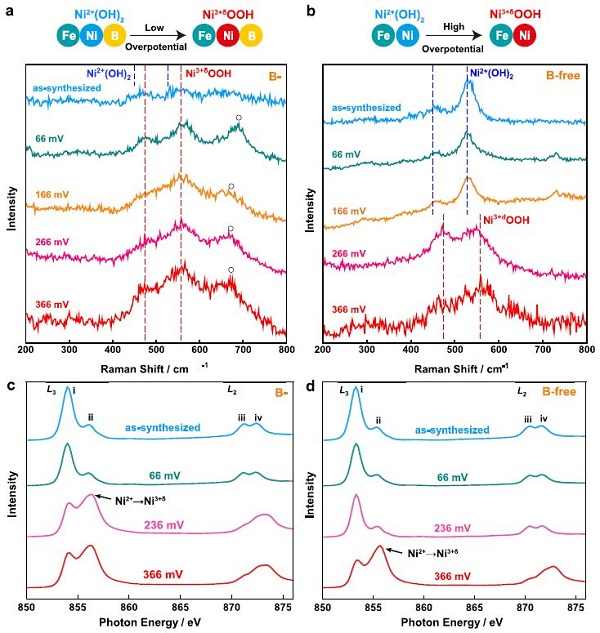
图2.NiFeB氢氧化物在不同过电位下的 (a,b) 拉曼光谱;(c,d) Ni L3,2边XAS光谱。
为了揭示B的作用,作者收集了NiFeB氢氧化物纳米片的拉曼光谱,以监测Ni在不同过电位下的氧化态转变(图2a)。NiFeB氢氧化物纳米片的光谱中没有明显的拉曼信号。当过电位增加到66 mV时,在~474和558 cm-1处出现了清晰的拉曼峰,对应于Ni3+δOOH。
当过电位增加到166、266和366 mV时,这些峰值保持不变。位于680 cm-1处的宽拉曼峰可以归因于NiFeB氢氧化物纳米片中的Fe-O。实验结果表明,在所有过电位下,NiFeB氢氧化物纳米片中的Ni均以Ni3+δOOH的形式存在。NiFeB氢氧化物纳米片在过电位为66 mV或更高时形成了具有电催化活性的Ni3+δOOH。
作者研究了超薄无B的NiFe氢氧化物纳米片在不同过电位下的拉曼光谱(图2b)。NiFe氢氧化物纳米片在449和527 cm-1处有明显的拉曼峰,这分别是由于Ni2+(OH)2中Ni2+ -OH和Ni2+-O键的振动引起的。
这些峰在过电位为0 ~ 166 mV的范围内保持不变,表明在此条件下纳米片中的Ni保持为Ni2+(OH)2,其OER活性不高。只有当过电位增加到266 mV或更高时,才会出现OER的活性Ni,即Ni3+δOOH。因此,在无B的NiFe氢氧化物纳米片中,形成有OER活性的Ni3+δOOH是一个更为困难的过程。
作者进一步利用XAS验证了NiFeB和NiFe氢氧化物纳米片中Ni在不同过电位下的氧化态转变(图2c, d)。测试前,纳米片保持在不同的过电位(66, 236和366 mV)。在Ni L3,2边XAS谱中,i和iii峰是由八面体晶体场中Ni 2p轨道上的电子跃迁到3d t2g轨道上产生的,ii和iv峰是由Ni 2p轨道上的电子跃迁到3d eg轨道上产生的。
ii/i峰强度比反映了eg和t2g轨道上的空穴比例,是衡量催化剂中Ni氧化态的指标。这些结果证实了B在促进Ni氧化态转变中的关键作用,与拉曼光谱显示的趋势一致。
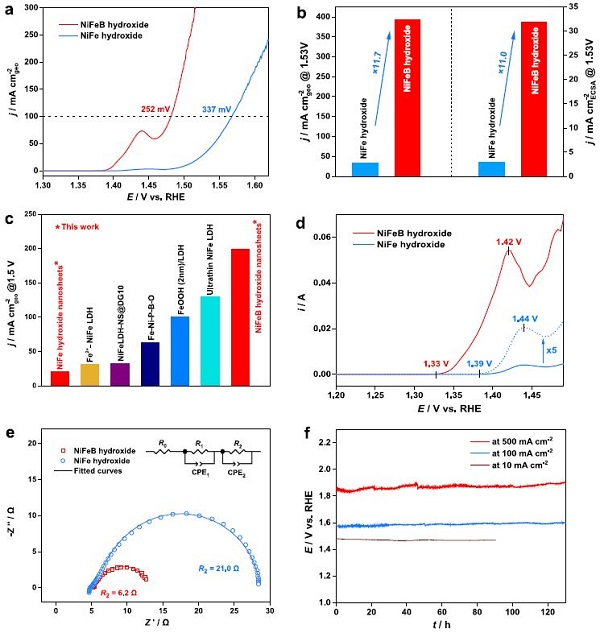
图3.电催化析氧性能比较。(a)OER极化曲线 (95% iR补偿)。(b) 不同催化剂在1.53 V下观察到的电流密度比较。(c) NiFeB氢氧化物纳米片与NiFe氢氧化物纳米片及典型非贵金属催化剂的OER活性比较。(d) 差分脉冲伏安曲线 (95% iR补偿)。(e) 在恒定电位为1.50 V vs. RHE时的奈奎斯特图。(f) 催化剂在电流密度为10、100和500 mA cmgeo−2下的时间电位曲线。
用线性扫描伏安法在O2饱和的1.0 M KOH中测量OER活性(图3a)。NiFeB氢氧化物纳米片的催化活性明显高于无B的NiFe氢氧化物纳米片。为了达到100 mA cmgeo-2的电流密度,NiFeB氢氧化物纳米片需要比NiFe氢氧化物纳米片(337 mV)低得多的过电位(252 mV)。NiFeB氢氧化物纳米片在1.53 V vs. RHE条件下,即过电位为300 mV时,电流密度为393 mA cmgeo-2,是NiFe氢氧化物纳米片(33.6 mA cmgeo-2)的11.7倍(图3b)。
就1.5 Vvs RHE下的电流密度而言,与文献报道的典型非金属催化剂相比,NiFeB氢氧化物是OER的首选催化剂(图3c)。通过差分脉冲伏安法(DPV)研究Ni氧化峰,进一步研究了Ni2+(OH)2→Ni3+δOOH跃迁的电化学过程(图3d)。NiFeB和NiFe纳米片的氧化峰起始电位分别为1.33和1.39 V。
因此,将B掺入到NiFe氢氧化物催化剂中,引起了Ni氧化峰的60 mV负移。B的峰位也发生了20 mV的负移。这一结果再次强调了B在低过电位下促进高氧化态Ni形成的作用,与Raman和XAS观察结果一致。值得注意的是,NiFeB氢氧化物纳米片的Ni氧化峰明显高于NiFe氢氧化物纳米片的Ni氧化峰,表明在B的促进下,NiFeB氢氧化物催化剂形成了更多的活性Ni3+δOOH物种。NiFeB氢氧化物催化剂具有较低的起始电位和较多的活性Ni3+δ物种,可能是其具有优异OER活性的原因。
为了更好地评价NiFeB氢氧化物纳米片的本征活性,作者检测了催化剂对ECSA归一化的OER电流密度(图3b)。LSV数据表明,NiFeB氢氧化物纳米片的性能优于无B的NiFe氢氧化物纳米片。在1.53 V下,NiFeB氢氧化物纳米片的电流密度为31.8 mA cmECSA-2,是NiFe氢氧化物纳米片(2.9 mA cmECSA-2)的11.0倍。
上述结果证实了B在提高NiFeB氢氧化物催化剂OER本征催化活性方面起到关键作用。图3e显示了催化剂在1.50 V下的Nyquist图。NiFe和NiFeB氢氧化物纳米片的电化学电荷转移电阻分别为21.0和6.2 Ω。NiFeB氢氧化物的电荷转移阻抗比NiFe氢氧化物低得多,这与NiFe催化剂的OER活性非常吻合。
单层NiFeB氢氧化物纳米片在OER中也表现出较高的稳定性。图3f显示了NiFeB氢氧化物纳米片在恒定电流密度为10、100和500 mA cm−2时的时间电位曲线。NiFeB氢氧化物纳米片维持电流密度所需的电势在130 h内保持稳定。
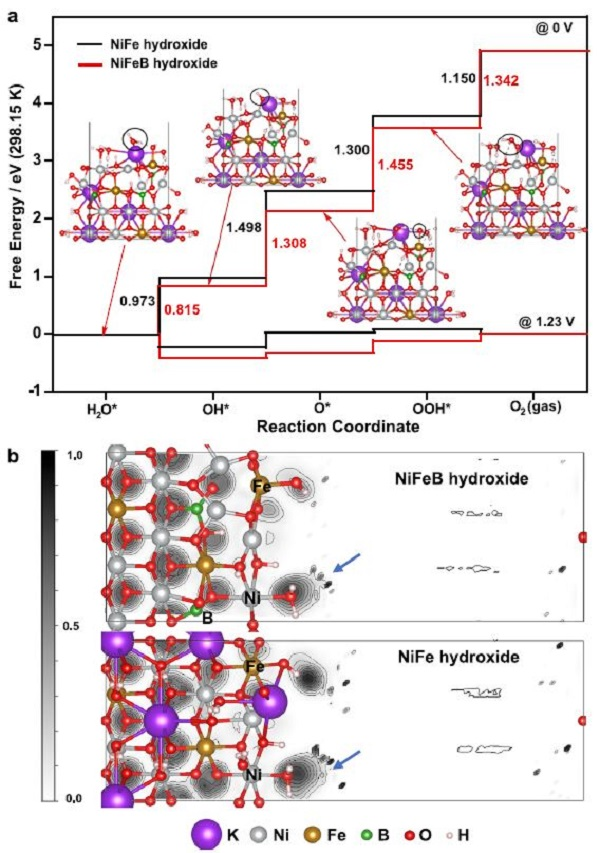
图4. DFT 计算。(a) NiFeB和NiFe氢氧化物电催化OER的自由能图。(b) NiFeB(上)和NiFe氢氧化物(下)的电子局域函数(ELFs)。
根据DFT计算,B的引入降低了OER的能垒。通过研究OER中间体的自由能来预测NiFeB氢氧化物和NiFe氢氧化物催化剂的OER活性(图4a)。考虑了由*H2O到O2的反应路径上有*OH,*O和*OOH中间体的四步反应路径。计算了两种电位(0和1.23 V)下的自由能,结果显示自由能沿反应坐标的变化趋势相似。
0 V时,NiFe氢氧化物形成*O (*OH→*O)为电位决定步骤(PDS),自由能差为1.498 eV,与前人结果一致。引入B后,NiFeB氢氧根上*O的生成能降至1.308 eV。而*OOH (*O→*OOH)的生成能增加到1.455 eV,*OOH形成了PDS。NiFeB氢氧化物的OER活性提高,说明了B在提高NiFeB氢氧化物催化剂OER活性中的关键作用。
DFT计算还表明,在NiFe氢氧化物中引入B后,Ni的氧化态升高。这些电子变化由ELFs表示(图4b)。采用灰度格式来阐明ELFs,其中0.0(白色)表示完全离域状态,1.0(黑色)表示完全局域状态。引入B后,Ni结合OH(如图中箭头所示)的电子局域化增强了,这为B附近Ni的氧化态增加提供了间接证据。
为了定量评估这些电荷,进行了Bader电荷分析,结果显示形式电荷最大值为+0.06。电子相互作用使B对Ni元素施加电子下沉效应,促进氧化态转变,有效降低了OER的起始电位,因此具有良好的催化活性。
总结
作者通过将NiFeB合金纳米粒子在碱性介质中水解,合成了单层NiFeB氢氧化物纳米片。TEM、XRD和XPS结果证实了MO6的单层形成(厚度约0.5 nm),未发生层间堆积。NiFeB氢氧化物纳米片在碱性电解质中表现出优异的催化活性和显著的长期稳定性。
只需要252 mV的过电位就可以达到100 mA cmgeo−2的电流密度,优于文献报道的大多数非贵金属OER催化剂。拉曼光谱、XAS和电化学DPV分析表明,OER活性与Ni2+向Ni3+δ的转变有关,在NiFe氢氧化物中加入B可有效降低Ni3+δ的电位。
更具体地说,DPV分析表明,将B加入到NiFe氢氧化物催化剂中,导致了启动Ni氧化态转变的电位发生了60 mV的负移。DFT结果表明,B增加了Ni的氧化态,改变了OER的速率决定步骤,降低了能垒,解释了实验结果。所有这些结果都支持了预想的假设,即B促进高氧化态Ni3+δ形成,生成OER所需的活性物种。
结果表明:(1)将B加入NiFe氢氧化物是制备高效OER催化剂的有效途径;(2) B的加入降低了Ni2+氧化为活性Ni3+δ基团所需的电位,降低了OER的起始电位,增强了OER的动力学;(3)在电催化OER过程中,缺电子的B可能起到了促进Ni2+氧化为Ni3+δ的作用。此研究为镍基催化剂的电荷工程提供了一个稳健的策略,在高性能电化学水裂解应用中实现高效的OER。
文献链接:
https://doi.org/10.1038/s41467-022-33846-0
审核编辑:刘清
-
氧化物半导体甲烷敏感元件详解2018-10-24 4942
-
氢氧化亚镍的性质2009-11-05 5094
-
片状氢氧化镍的合成及电化学性能研究浅谈2009-12-07 2646
-
为什么氢氧化锂价格后期走势变化这么大2018-09-18 2174
-
LME确定选用氢氧化锂作为锂期货合约标的,并于明年上半年推出2020-10-23 4038
-
电池材料碳酸锂与氢氧化锂的区别2021-03-18 14305
-
如何解决氢氧化钠罐体腐蚀问题2021-05-28 2645
-
硅晶片在氢氧化钾、TMAH和EDP溶液中的蚀刻速率2022-04-26 5317
-
栅极氧化物形成前的清洗2022-06-21 3190
-
二维石墨烯多层纳米限域金属有机框架获得超高析氧活性2022-11-10 2305
-
硅在氢氧化钠和四甲基氢氧化铵中的温度依赖性蚀刻2023-05-29 3666
-
不同行业对氢氧化铝阻燃剂都有些什么要求?2023-07-20 2001
-
京朗仕特氢氧化钙化验设备检测方法升级了2025-04-01 783
全部0条评论

快来发表一下你的评论吧 !
