

揭示卤素掺杂Sn基催化剂促进CO2电还原制甲酸盐原因
描述
全文速览
非金属掺杂的Sn基催化剂展现出优异的电化学催化CO2还原活性。然而,非金属掺杂原子在电化学还原电位下的流失以及相应的催化剂结构的动态演变过程仍然亟待厘清。针对这一挑战,新加坡南洋理工大学刘彬教授,新加坡国立大学Sibudjing Kawi教授和苏州科技大学杨鸿斌教授合作,通过简单的室温水解反应制备卤素掺杂的Sn基催化剂,并展现出优异的电化学催化CO2 制甲酸盐活性。
原位拉曼结果证实了卤素(氯)掺杂在电化学还原电位下的稳定存在和SnO2到Sn的动态转变过程。随后的原位红外和第一性原理计算结果表明,残留的卤素掺杂可以有效调控Sn活性位点的电子结构和对甲酸盐中间产物的吸附能和反应路径的能垒,从而展现出优异的甲酸盐选择性。
背景介绍
由于其对甲酸/甲酸盐中间产物适中的吸附能,Sn基催化剂被广泛应用在电催化CO2还原产甲酸的反应中。通过对其进行非金属掺杂,催化的活性和选择性还可以被进一步提高。然而,不同于金属掺杂和合金化策略,非金属掺杂原子在CO2电化学还原电位下非常容易被还原并浸出从而影响催化剂性能。
此外,常见的非金属掺杂策略通常需要额外的处理步骤,从而提高了制备过程的复杂程度。因此开发一种简便的制备非金属掺杂Sn基催化剂并研究其在电化学还原电位下的动态演变过程对深入理解非金属掺杂对Sn基催化剂的调控原理和机制就显得尤为重要。
研究内容
1) 卤素掺杂的Sn基催化剂是在室温下通过简单的水解方法制备的。以Cl掺杂为例,在Ar和O2气氛中,在室温下通过SnCl2与H2O的水解反应分别制备Cl掺杂Sn和未改性Sn催化剂(图1a)。图1b-d对比了二者的XRD图谱和SEM能谱结果。由于Sn3O(OH)2Cl2合成过程中的惰性气氛(Ar),Sn3O(OH)2Cl2中的Sn价态保持在+2价,因此在XPS图谱中表现出了向低结合能方法的偏移。通过X射线吸收光谱(XAS)进一步研究了所制备催化剂的电子结构。Sn3O(OH)2Cl2在1.65 Å左右的峰可归因于Sn-O和Sn-Cl键,因此明显不同于SnO2催化剂中的Sn-O键(~1.58 Å)。此外,与SnO2相比,Sn3O(OH)2Cl2在1.60 Å附近的峰值较低,这与Sn3O(OH)2Cl2中较低的Sn-O配位数一致。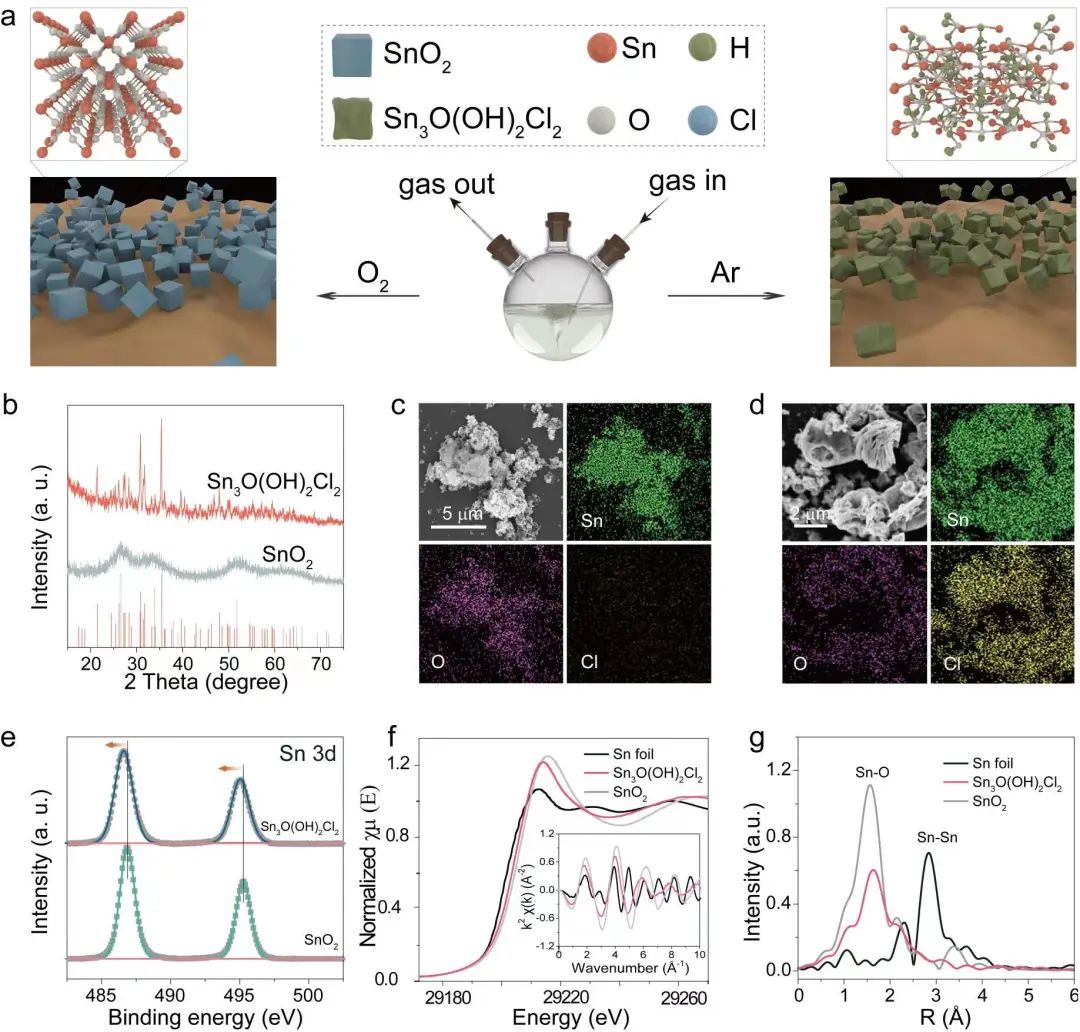
图1 (a) SnO2和Sn3O(OH)2Cl2催化剂制备示意图。(b) XRD图谱。(c) SnO2和(d) Sn3O(OH)2Cl2的SEM图像和相应的EDX图。(e) SnO2和Sn3O(OH)2Cl2的Sn3d谱,(f) Sn K边X射线吸收近边结构(XANES)。插图为k空间EXAFS谱。(g) Sn箔、SnO2和Sn3O(OH)2Cl2的扩展X射线吸收精细结构光谱(FT-EXAFS)。
2)通过线性扫描伏安法(LSV)在三电极系统中评估所制备催化剂的CO2RR性能。如图2a所示,SnO2和Sn3O(OH)2Cl2在CO2饱和KHCO3电解质中表现出比在Ar饱和KHCO3电解质中更高的电流密度,表明Sn基催化剂优异的CO2RR性能。此外,与SnO2相比,Sn3O(OH)2Cl2在CO2饱和电解质中表现出更小的过电位和更大的电流密度。CO2RR产物通过在线气相色谱(GC)和高效液相色谱(HPLC)进行量化计算。在最终的CO2RR产品中仅检测到气态CO、H2和液态甲酸盐。图2b显示了在不同电位下的CO、H2和甲酸盐的法拉第效率(FE)。
随着还原电位的增加,Sn3O(OH)2Cl2上的甲酸盐FE增加,达到最大值96.1±2.3%(-0.9 V vs. RHE)。此时的甲酸盐电流密度为62.4 mA cm-2(图2c),远高于SnO2 在-0.9 V的62.3±3.4%的甲酸盐选择性和14.5 mA cm-2的甲酸盐电流密度。
更重要的是,Sn3O(OH)2Cl2上的甲酸盐FE可以在-0.9至-1.2V的宽电位窗口内保持高于93%的水平。除了活性和选择性外,Sn3O(OH)2Cl2催化剂还表现出优异的催化稳定性。在-0.9 V vs. RHE下连续反应35小时后,电流密度几乎保持不变,约为29 mA cm-2,而反应结束时甲酸盐FE仍然保持为90%(图2d)。与之前发表的Sn基催化剂相比,我们报道的Cl产则的Sn催化剂在电流密度和产物选择性方面表现出优异的性能。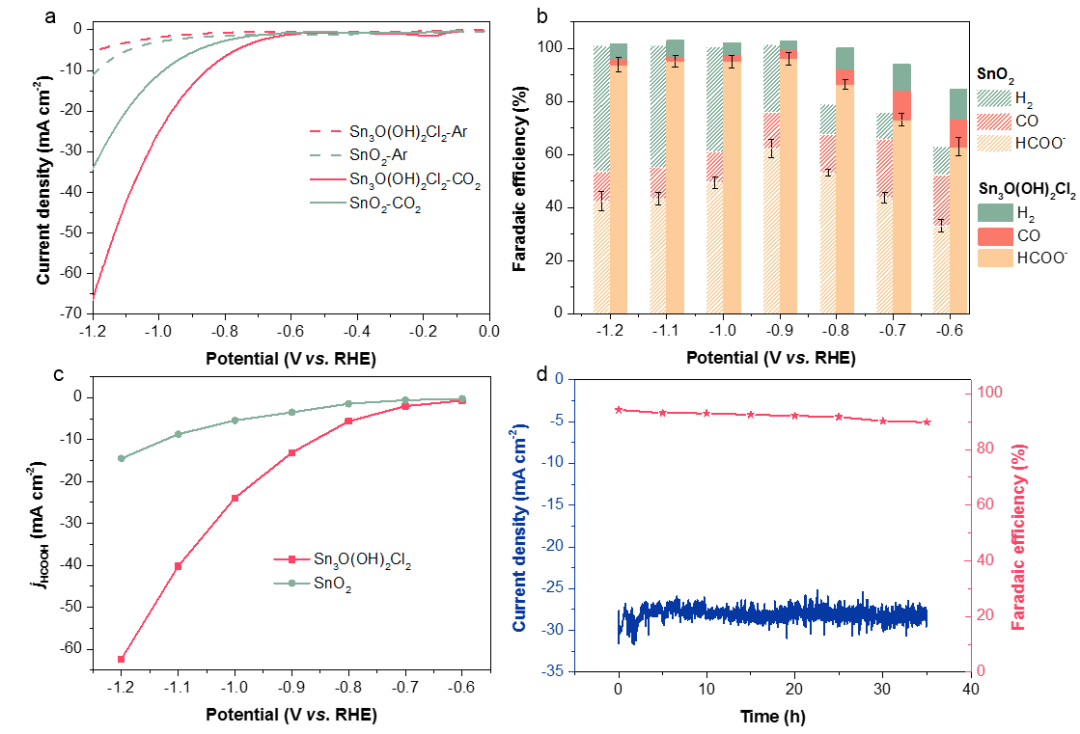
图2 (a) 在Ar和CO2饱和的KHCO3电解质中的Sn3O(OH)2Cl2和SnO2的LSV曲线。(b) Sn3O(OH)2Cl2和SnO2的CO2RR法拉第效率。(c) Sn3O(OH)2Cl2和SnO2的甲酸盐电流密度。(d) 稳定性测试。 3)通过XPS研究了SnO2和Sn3O(OH)2Cl2在CO2RR后Sn的价态(图3a-c)。考虑到金属Sn在空气中很容易被氧化,因此在收集Sn光谱之前进行Ar+溅射以去除氧化层。在Sn 3d XPS光谱中可以清楚地看到Sn0信号,证实了在CO2RR过程中SnO2和Sn3O(OH)2Cl2的还原。
此外,CO2RR后Sn3O(OH)2Cl2中Sn0的百分比明显小于CO2RR后SnO2中的Sn0(12.3% vs.19.3%),表明Sn和Cl之间可能存在电子转移。在O 1s和Cl 2p的XPS光谱中也可以观察到CO2RR后催化剂的还原和Sn3O(OH)2Cl2中Cl的保留(图3b和c)。考虑到Sn3O(OH)2Cl2催化剂中Cl的存在对其电子结构和催化性能起着重要的调节作用,基于XPS的结果研究了CO2RR过程中Sn3O(OH)2Cl2中Cl含量的演变。具体而言,CO2RR前后的Cl %分别为22.4%和7.8%,表明在还原电位下从Sn3O(OH)2Cl2转化为Cl掺杂Sn的过程中可能发生Cl浸出。
此外,长时间稳定性测试后的样品中,Cl %经计算为7.3%。使用离子色谱法进一步定量研究了CO2RR测试过程中和长期稳定性测试后电解液中Cl-的含量。几乎没有变化的Cl特征峰证实了Cl掺杂在CO2RR过程中可以很好地保留在Cl-Sn催化剂中。
为了进一步探究催化剂在CO2RR过程中的动态结构演变,进行了原位拉曼光谱测试。由于所制备的SnO2样品结晶度较差(图1b),SnO2特征振动峰(A1g、B2g和Eg)并未出现在50至250 cm-1的波数范围内。然而,随着外加还原电位的增加,可以检测到位于191 cm-1附近的Sn-Sn振动信号,表明SnO2还原形成金属Sn。
另一方面,Sn3O(OH)2Cl2在开路电位的拉曼光谱中位于133和227 cm-1附近的两个谱带可归因于弯曲振动ν5(Cl-Sn-Cl)和对称拉伸ν2(Sn-Cl)。这些带的强度随着还原电位的增加而逐渐增加,表明在CO2RR过程中Cl可以保留在Sn3O(OH)2Cl2中。
Sn-Cl和Cl-Sn-Cl强度的增加可以用CO2RR过程中O元素的损失来解释。该结果也与在CO2RR后观察到的Sn-Cl键百分比从16.28%增加到48.17%一致。因此,上述非原位TEM/EDX、XRD、XPS和原位拉曼光谱测试系统研究了催化剂在CO2RR过程中的结构演变,即SnO2和Sn3O(OH)2Cl2在CO2RR中被还原,而Cl将保留在Sn3O(OH)2Cl2中。
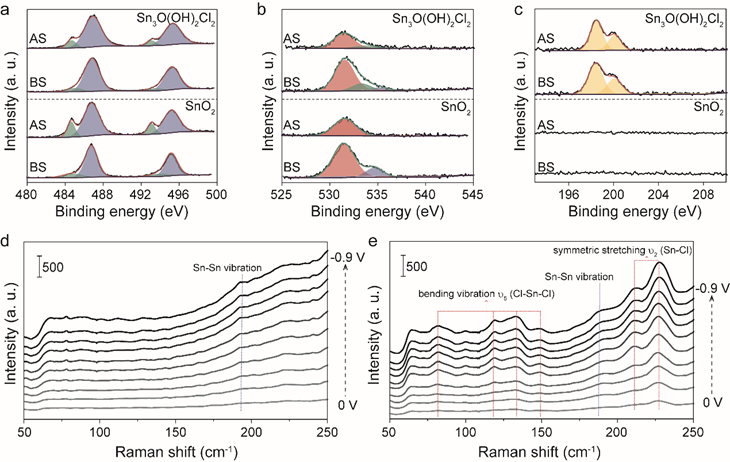
图3 Ar+溅射前后CO2RR后SnO2和Sn3O(OH)2Cl2的XPS光谱:(a) Sn 3d,(b) O 1s,和(c) Cl 2p。(d) SnO2和(e) Sn3O(OH)2Cl2在不同还原电位下的原位拉曼光谱。
4)在原位红外测试中,位于1410cm-1的吸收峰可归因于双齿*OCHO中的O-C-O振动,这在SnO2(图4a)和Sn3O(OH)2Cl2的光谱中均有检测到(图4c)。而在2852和2975 cm-1波数范围内的C-H伸缩振动被认为是CO2RR中产生甲酸/甲酸盐的特征吸收。值得注意的是,Sn3O(OH)2Cl2上的*C-H强度比SnO2上的强得多。
另一方面,*CO被认为是CO2RR形成CO的关键中间体,出现在1863 cm-1附近。与Sn3O(OH)2Cl2上记录的弱*CO吸收峰相比,SnO2上的*CO吸收强度要强得多。此外,对于SnO2催化剂,*CO吸收峰开始出现在-0.4 V vs.RHE,而对于Sn3O(OH)2Cl2催化剂,只有当施加的电位比-0.8V更负时才会出现,表明在SnO2形成*CO是热力学更有利的。
为了检查反应的稳定性,将电位固定在-0.9 V vs. RHE,并记录原位ATR-SEIRAS光谱。如图4b和d所示,SnO2和Sn3O(OH)2Cl2上的CO2RR特征峰保持稳定,表明良好的催化稳定性。
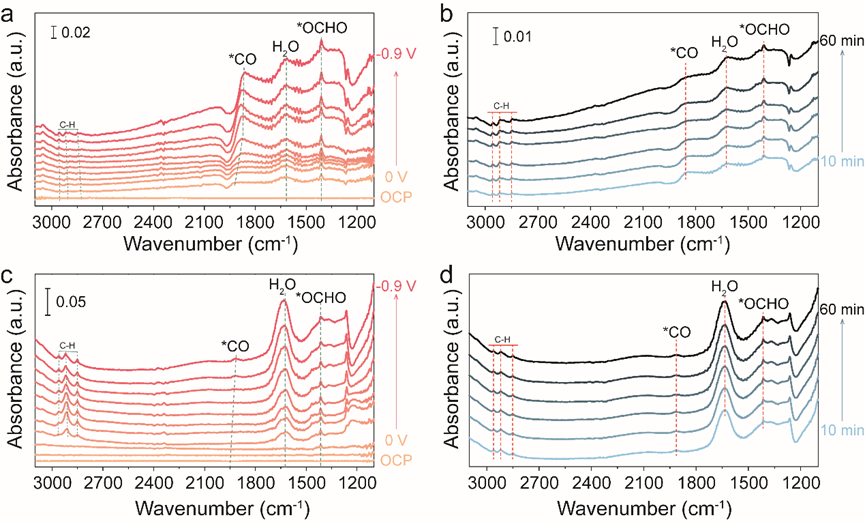
图4 在CO2饱和电解质中不同还原电位下记录的ATR-SEIRAS光谱:(a) SnO2和(c) Sn3O(OH)2Cl2。在-0.9 V vs. RHE下保持不同时间的ATR-SEIRAS光谱:(b) SnO2和(d) Sn3O(OH)2Cl2。 除了Cl,其他卤素元素如F、Br和I也可以通过类似的水解方法掺杂到Sn中。
与金属Sn相比,所有这些催化剂在CO2RR中都表现出更好的催化性能。掺入卤素的Sn的电流密度与不同的CO2吸附能力有关,这可以通过氧化LSV扫描得到验证。另一方面,不同的甲酸盐选择性可以由甲酸盐产生的能垒来解释。因此,实验和理论证据证明卤素改性可以作为提高电化学CO2RR性能的普适性策略。
总结和展望
综上所述,我们开发了一种简便的水解策略,可在室温下合成用于电化学CO2RR的卤素改性Sn催化剂。合成后的Sn催化剂在CO2工作电位下被还原为金属Sn位点以催化CO2RR。将卤素掺杂到Sn中可以诱导电子从Sn转移到卤素,这有效地提高了Sn的价态,从而促进CO2RR形成甲酸盐,同时抑制CO2RR产CO和HER的竞争反应。
审核编辑:刘清
- 相关推荐
- 热点推荐
- 催化剂
-
反向电子转移!双-单原子催化剂助力CO2光还原2023-08-29 3233
-
大面积二维Cu2Te垂直阵列催化剂助力CO2电还原2023-07-17 2575
-
CO2辅助生成富含晶界的Cu催化剂实现高效CO-CO偶联2023-03-17 2114
-
依赖于尺寸和载体的Ptn/X-石墨烯催化剂(X = C、B、N)减弱CO中毒2023-01-11 2083
-
分子催化剂助力酸性条件下的CO2还原2023-01-10 3090
-
金属簇催化剂的CO2转化反应性和循环性2023-01-09 1656
-
如何更好地报道CO2电还原的性能?2023-01-08 2869
-
CO2转化为C2产物的高效光催化剂的设计研究2022-11-25 4650
-
通过Cl-吸附在Ag HF电极上使CO2电还原为CO的效率2022-10-18 2481
-
蜂窝状多孔结晶异质电催化剂实现高效的CO2吸附/活化2022-09-30 4293
-
生成CO的催化剂与Cu之间的相互作用2022-08-22 4048
-
一类新的钌基催化剂,采用原位制备技术2022-08-13 3063
-
氧气还原反应催化剂的制作及性能研究2021-08-09 891
-
碱性醇类燃料电池新型催化剂的研究2011-03-11 1987
全部0条评论

快来发表一下你的评论吧 !
