

一文简析磷表面氧化提高储锂性能
描述
01
研究背景
由于具有较高的理论容量(2596 mAh g–1)和适当的锂化电势(∼0.7 V vs Li+/Li),磷被认为是锂离子电池最有前途的负极材料之一。然而,它具有大的体积变化(∼300%),低电导率(∼10–14 S cm–1)和不稳定的固体电解质界面(SEI)等缺点,导致电化学循环衰减迅速。
为此,研究人员采用了各种策略来解决这些问题,包括结合各种碳框架来实现P颗粒的纳米限制,引入导电聚合物涂层以缓解体积膨胀,以及应用功能电解质来构建高弹性SEI膜。尽管上述问题已经基本解决,但磷在制备过程中容易被氧化。因此,磷基负极材料的粉末合成、浆料制备和电极烘焙对避免氧化有很高的要求。
02
成果简介
近日,天津大学孙洁教授和贵州振华电子化工有限公司Qianxin Xiang在Nano Letters上发表了题为“Surficial Oxidation of Phosphorus for Strengthening Interface Interaction and Enhancing Lithium-Storage Performance”的论文。该论文展示了一种原位预氧化策略,以在磷颗粒上构建功能性氧化层。氧化层不仅作为保护层延长了磷负极在空气中的储存时间,还碳化了N-甲基吡咯烷酮和聚偏氟乙烯,加强了磷颗粒与粘结剂之间的界面相互作用。氧化物层进一步诱导形成具有高锂离子电导率的稳定固体电解质界面。在100次循环后, 氧化的P-CNT保持1306 mAh g–1的高比容量,容量保持率高达89%,远高于原始P-CNT(17.1%)。原位氧化策略简单易行,有利于磷基负极的实际应用。
03
研究亮点
(1)在浆料制备过程中,氧化磷可以碳化N-甲基吡咯烷酮(NMP)溶剂和聚偏氟乙烯(PVDF)粘结剂,加强磷颗粒与粘结剂之间的界面相互作用。
(2)氧化层可以作为保护层,延长磷负极在空气中的储存时间。
(3)氧化物层可以进一步诱导形成具有高锂离子电导率的稳定SEI。
04
图文导读
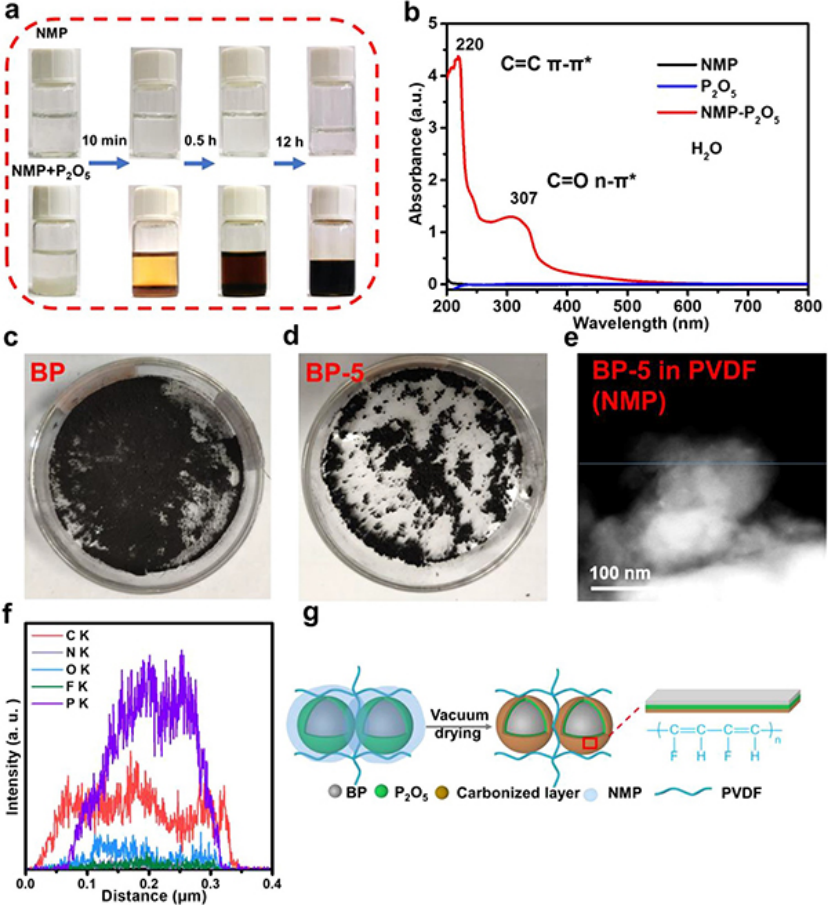
图 1、(a)分别在真空干燥10分钟、0.5小时和12小时后,仅NMP溶剂和含有P2O5的NMP的光学图像。(b)含有P2O5的NMP,仅含有NMP,仅含P2O5紫外可见光谱。(c)原始BP粉末和(d)氧化BP粉末图像。(e-f)BP-5在含有3%PVDF的NMP中的STEM图像和线扫描结果。(g)浆料制备过程中碳化层形成的示意图。
在浆料制备过程中,NMP和PVDF粘结剂与磷氧化物发生碳化反应,因为磷氧化物是强脱水和碳化剂,而NMP和PVF是含有碳、氢、氮和氟的有机物质。为了验证这一点,将P2O5添加到NMP中,将其置于80°C的真空干燥箱中。当P2O5在10分钟后添加到NMP中时,溶液的颜色呈现为浅黄色(图1a)。当时间分别延长到0.5小时和12小时时,溶液的颜色变得越来越深。然而,即使在相同条件下12小时后,纯NMP溶剂的颜色仍保持无色和透明。
将反应12小时的纯NMP和NMP+P2O5溶液干燥以完全去除NMP,并分别在等体积的去离子水中重新溶解,以进行UV-vis光谱测试(图1b)。NMP+P2O5的紫外-可见光谱分别在220和307 nm附近显示出两个不同的吸收峰(图1b)。220 nm附近的吸收峰归因于C═C键的π–π*电子能级跃迁,而307 nm处的吸收峰归因于C═O键的n–π*跃迁。
为了消除残余P2O5和NMP的干扰,将P2O5粉末作为对照样品添加到去离子水中,以进行UV-vis光谱测试。P2O5(蓝线)和NMP(黑线)的对照样品在220和307 nm处没有出现类似的吸收峰(图2b),表明C═C键来源于NMP的碳化反应。
为了进一步验证PVDF和NMP与P表面自然形成的氧化物层碳化的反应,将在室温下氧化5天的黑磷(BP-5)与新鲜黑磷(BP)进行比较。BP表现出均匀和松散的分布,而BP-5表现出严重的团聚现象(图1c,d)。将这两个样品分别加入含有3%PVDF的NMP和NMP中,搅拌过夜,然后用环己烷洗涤3次,以排除残余PVDF和NMP对样品的影响。
之后,将所得产物过滤并在真空中干燥以进行STEM线扫描。对于BP-5在NMP-3%PVDF溶液中的情况(图1e,f),分别有来自NMP的C和N元素、来自PVDF的F元素以及来自氧化磷的P和O元素的信号。C元素的分布比P和O元素的分布更广,表明NMP和PVDF在黑磷表面会发生碳化反应。
因此,浆料制备过程中碳化层形成的示意图如图1g所示。碳化反应可以增强磷与粘结剂的相互作用,有利于降低界面阻力,提高循环稳定性。
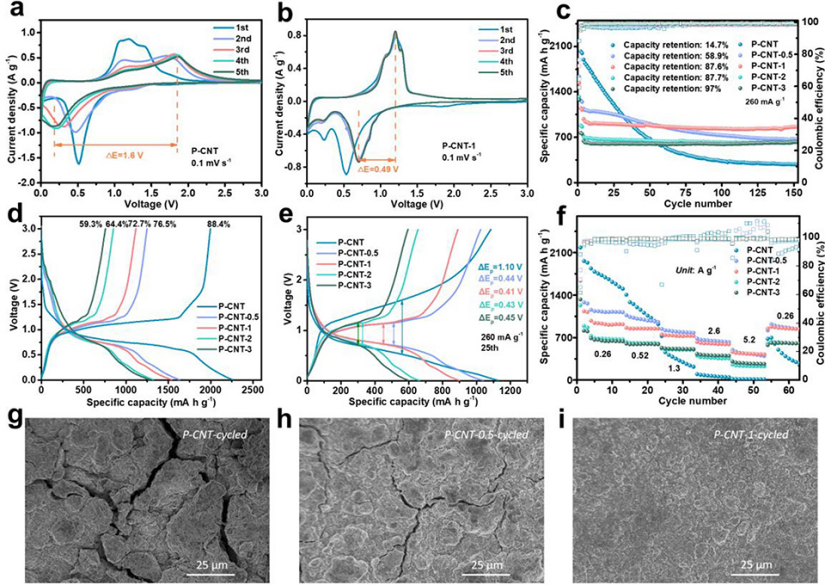
图 2、(a)P-CNT和(b)P-CNT-1负极的CV曲线,扫速为0.1 mV s–1。(c)长循环稳定性。(d)50 mA g–1下的初始充放电曲线。在260 mA g–1下,(g)P-CNT、(h)P-CNT-0.5和(i)P-CNT-1电极25次循环后的SEM图像。
图2a显示,在第一次阴极扫描中,原始P-CNT负极在~1.16 V处显示了一个宽的不可逆峰,这是由于电解质的不可逆分解形成SEI。0.52 V的另一个还原峰对应于从P到LixP化合物的逐步锂化过程(x=1–3)。在第一次阳极扫描中,1.19V处出现阳极峰,这源于LixP的逐步脱锂过程。
在随后的循环中,阴极峰逐渐移动到低电压,而阳极扫描的峰位置逐渐移动到高电压,峰面积减小,表现出递增的极化和较差的循环稳定性。而P-CNT-1(图2b)负极由于其小的过电位、随后的CV曲线高度重叠,表现出高的可逆性。
图2c显示,原始P-CNT在第一次循环中表现出2025.1 mAh g–1的高初始比容量,但在150次循环后,容量逐渐下降至279 mAh g-1,保留率仅为14.7%。P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的初始放电比容量分别为1632.4、1536.6、1322.2和1278.1 mAh g–1。由于氧化层的产生导致活性磷的一定损失,初始容量随着氧化期的增加而逐渐减小。
此外,初始库仑效率(ICE)从原始电极的88.4%逐渐降低到P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的76.5%、72.7%、64.4%和59.3%(图2d)。在260 mA g–1下循环150次后,P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的容量保持率分别增加到58.9%、87.6%、87.7%和97%。图1e显示,P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的充放电电压极化(ΔEp)分别为0.44、0.41、043和0.45 V,明显小于原始P-CNT(1.10 V),表明氧化样品具有降低的内阻。
倍率性能如图2f所示。尽管P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的初始放电容量显著低于原始P-CNT,但当提高电流密度时,前者的容量衰减较小。因此,表层氧化物层的存在有利于提高倍率性能。
通过SEM表征了25次循环后P-CNT、P-CNT-0.5和P-CNT-1电极的表面形貌。由于体积变化较大,原始P-CNT电极中存在严重裂纹(图2g)。对于P-CNT-0.5电极(图2h),尽管电极表面的完整性得到了改善,但由于磷颗粒的表面氧化不足,仍然可以观察到一些微小的裂纹。
相反,P-CNT-1电极(图2i)保持致密和光滑,没有明显断裂。P-CNT、P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的ICE随着氧化时间的增加而逐渐减少(图2d)。这种不可逆的容量损失主要来自SEI形成的消耗。
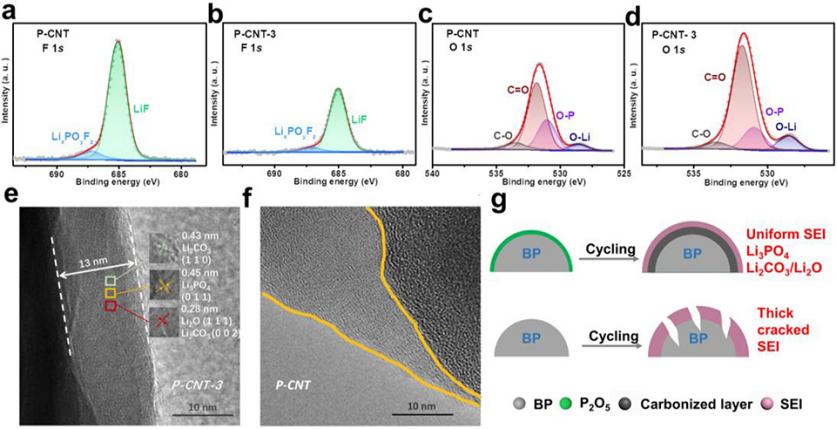
图 3、150次循环后,P-CNT和P-CNT-3电极的高分辨率F 1s(a)、(b)和O 1s(c)、(d)XPS光谱。(e)P-CNT和(f)P-CNT-3电极的高分辨率TEM图像。(g)SEI形成过程示意图。
150次循环后,P-CNT和P-CNT-3的F 1s和O 1s XPS光谱如图3a–d所示。F 1s光谱可以拟合为LiF(685.1eV)和LixPOyFz(687.1eV),它们是电解质分解的副产物。与P-CNT(图3a)相比,P-CNT-3(图3b)的F 1s光谱中LiF和LixPOyFz峰面积更低,表明氧化物层可以有效减少电解质与活性磷的副反应。O1s可以拟合成O–Li(528.6 eV)、O–P(531.0 eV)、C═O(531.8 eV)和C–O(533.5 eV)键。O 1s光谱中的O–Li信号主要对应于SEI中具有优异离子电导率的Li3PO4、Li2O和Li2CO3。P-CNT-3的O1s光谱中O–Li键的峰面积明显大于原始P-CNT,表明P-CNT-3中产生了高含量的Li3PO4、Li2O和Li2CO3。
通过TEM表征了150次循环后P-CNT和P-CNT-3电极的SEI形态(图3e,f)。P-CNT-3在循环后呈现出均匀的SEI层,约13nm,其主要包含Li3PO4、Li2CO3和Li2O。而原始P-CNT的SEI层呈现出不均匀的分布。图3g显示,富含Li3PO4和Li2CO3的SEI不仅有助于缓解体积膨胀,而且提高了反应动力学。
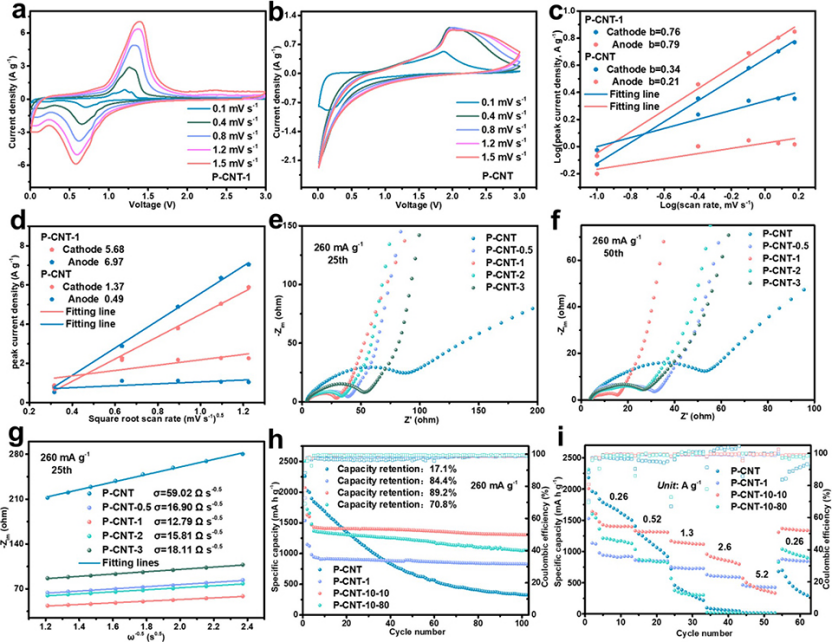
图 4、在0.1至1.5 mV s–1不同扫速下,LIB的(a)P-CNT-1和(b)P-CNT的CV曲线。(c)阴极和阳极峰的Log(峰电流)–Log(扫速)曲线。(d)P-CNT-1和P-CNT负极阴极和阳极峰的Ip/ν1/2曲线。P-CNT, P-CNT-0.5, P-CNT-1, P-CNT-2和P-CNT-3电极在循环(e)25,(f)50圈之后的EIS谱,以及循环25圈后角频率均方根倒数与低频下−Zim的关系。P-CNT、P-CNT-1、P-CNT-10-10和P-CNT-10-80的(h)长循环稳定性和(i)倍率性能。
进行CV测试以研究不同扫速的动力学因素(图4a,b)。方程1和2用于研究反应动力学过程。

其中,i表示峰电流(mA),ν表示扫速(mV s–1),a和b表示可变参数。
P-CNT-1负极的阴极(0.76)和阳极(0.79)峰b值明显大于P-CNT负极(0.34和0.21)(图4c),表明前者具有更好的动力学性能。此外,利用Randles–Sevcik方程(方程式3)研究扩散系数。
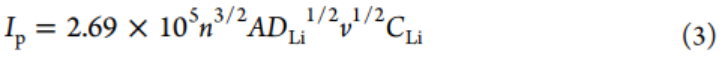
其中Ip、n、A、DLi、CLi和v表示峰电流、电荷转移数、电极面积、Li离子扩散系数、Li离子浓度和扫速。P-CNT-1和P-CNT电极的DLi可以通过线性拟合Ip与v1/2来评估。P-CNT-1电极的阳极和阴极峰的Ip/v1/2值分别为6.97和5.68(图4d),远大于P-CNT的值(0.49和1.37),表明P-CNT-1电极在原始P-CNT上具有更快的Li离子扩散。
分别在260 mA g–1下进行1次、25次、50次和100次循环后,获得了P-CNT、P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3电极的EIS光谱。如图4e、f所示,氧化样品(P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3)在1、25、50和100次循环后的电荷转移电阻明显小于P-CNT,表明氧化物层可以有效地降低界面电荷转移电阻。
方程4和5用于计算Li+扩散系数。
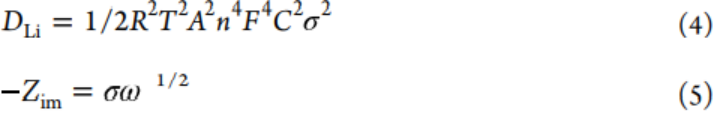
其中,气体常数、绝对温度、电极面积、法拉第常数和频率分别用R、T、A、F和ω表示。电池阻抗的虚部和低频区域的Warburg因子分别用−Zim和σ表示。因此,利用由−Zim与ω-1/2拟合得到的斜率Warburg系数σ来评价离子电导率。所有氧化样品的σ值均低于原始P-CNT的σ值,无论其是否经过1、25、50和100次循环(图4g),并且P-CNT-1在四个氧化负极中具有最小的σ,表明其具有优异的离子传导性和中等厚度的SEI。
为了探索氧化层对P颗粒的保护作用,将P-CNT电极置于10%低湿度环境中1天,以在电极表面形成氧化物层。之后,将氧化电极分别置于10%低湿度(定义为P-CNT-10-10)或80%高湿度(定义P-CNT-10-80)环境中再放置1天。P-CNT-10-10的电极提供1306 mAh g–1的比容量,并在100次循环后,保持89.2%的容量保持率(图4h)。
P-CNT-10-80的电极表现出较差的循环性能,因为当其置于高湿度条件下时,氧化层由于吸附的水而被破坏。P-CNT-10-10的倍率性能高于P-CNT和P-CNT-10-80。P-CNT-10-10-10的容量和循环性能高于P-NT-1,表明电极氧化法明显优于粉末氧化法。
05
总结与展望
为了解决磷负极体积膨胀大、SEI层不稳定、空气和水稳定性差等问题,本工作提出了在磷表面原位包覆氧化层的策略。氧化层不仅可以起到保护的作用,延长磷负极在空气中的储存时间,还可以碳化NMP和PVDF,形成碳化层,加强磷颗粒和粘结剂之间的界面相互作用。生成的氧化层可以进一步诱导形成稳定、均匀和高Li+电导率的SEI。
氧化的P-CNT保持1306 mAh g–1的高比容量,100个循环后的容量保持率为87%,远高于原始P-CNT(17.1%)。这种原位氧化法简单,没有额外的材料或加工成本,有望实现工业生产。
审核编辑:刘清
-
掺杂氧化镍锰钴锂材料的动力型锂离子电池2011-03-04 1991
-
陶瓷隔膜氧化铝-提高电池安全性能2014-04-23 3811
-
锂电池正极材料锂锰氧化物的改性与循环寿命2009-11-03 535
-
氧化皮除磷设备适合清除哪些锻件表面氧化皮2018-09-17 492
-
氧化皮清洗机是一款可改善五金工具表面质量的设备2021-03-02 945
-
未来十年磷酸铁锂将取代锂锰钴氧化物成为固定式能量储能化学2020-08-26 888
全部0条评论

快来发表一下你的评论吧 !
