

PtNi纳米合金可与Ni单原子发生原子置换
描述
研究背景
多相金属催化剂在化工生产、环境保护和可持续能源生产中发挥着重要作用。为了实现催化活性金属的有效利用,金属物种通常被设计为高度分散的形式,如金属纳米颗粒、纳米团簇和单原子形式。探索催化活性金属物种在载体上的转化途径对于制备高效催化剂、研究催化剂如何失活以及废旧催化剂的再生至关重要。目前,烧结和再分散是多相催化剂中金属活性组分的两种主要转化方式,分别代表金属活性物质在载体上的自下而上和自上而下的转换。
成果简介
鉴于此,清华大学李亚栋院士与北京师范大学李治教授(共同通讯作者)等发现,PtNi纳米合金中的Ni与ZIF-8衍生氮掺杂碳(Zn1-CN)载体上的Zn在高温下发生了金属原子交换,生成PtZn纳米晶和Ni单原子(Ni1-CN)。(PtZn)n/Ni1-CN多位点催化剂在CO2还原反应(CO2RR)中表现出较低的CO2质子化和CO脱附能垒,大大提高了CO2RR的催化性能。
研究亮点
1、发现了一种新型金属催化剂,通过金属原子在单原子和纳米合金之间的交换作用,形成了一套新的纳米合金和单原子;
2、PtNi纳米合金可与Ni单原子发生原子置换,这种独特的原子置换转化也适用于其他金属合金,如PtPd等。
图文介绍
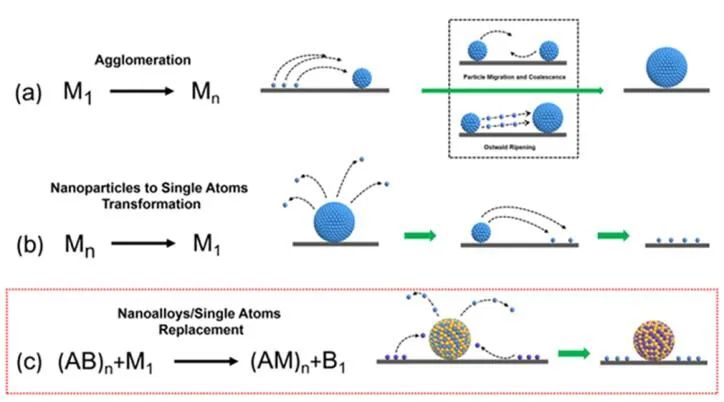
图1单原子和纳米粒子之间的转化途径。(a)单个原子凝聚成纳米粒子;(b)纳米颗粒再分散(单原子化)形成单个原子;(c)单原子和纳米合金之间的原子替换。
颗粒迁移/聚结和奥斯特瓦尔德熟化是金属烧结的两种主要机理。纳米粒子迁移/聚结表示纳米颗粒的移动和结合会形成更大的聚集体,奥斯特瓦尔德熟化表示原子级物种会从较小的纳米颗粒迁移到较大的纳米颗粒(图1a)。与烧结相反,再分散减小了金属活性组分的尺寸,增加了多相催化剂表面原子的比例。
目前,金属再分散可以实现金属纳米颗粒或大块金属到金属单原子的转化(单原子化)(图1b)。再分散/单原子化通常会显著提高催化性能,这也是实现废旧催化剂再生的重要途径。作者发现了一种全新的多相金属催化剂转化模式,即单原子与纳米合金之间的原子置换,即通过单原子与纳米合金之间的原子置换,形成单原子与纳米合金的新组合(图1c)。
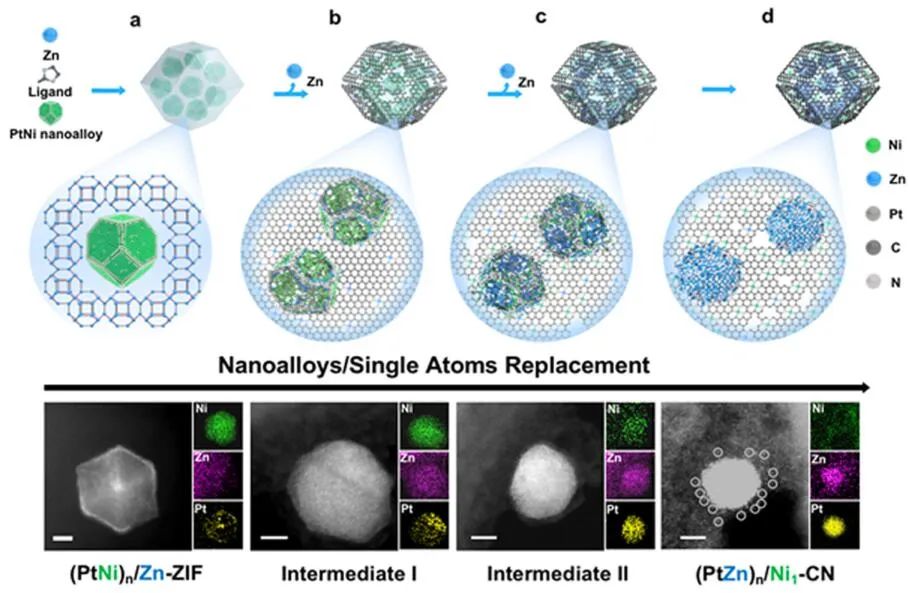
图2纳米复合材料的原子置换及结构表征。(a-d)(PtNi)n/Zn-ZIF(a)、中间体I(b)、中间体II(c)、(PtZn)n/Ni1-CN(d)的AC-STEM图像及其对应的EDS元素映射图。
在含有分散的PtNi纳米合金的Zn(NO3)2溶液中加入2-甲基咪唑溶液,制备了PtNi纳米合金/ZIF-8纳米复合材料。ZIF-8纳米晶体在PtNi纳米合金周围生长,生成(PtNi)n/Zn-ZIF纳米结构。EDS元素映射和元素线扫描显示,Zn作为ZIF-8的组成元素均匀分布在纳米复合材料中,而Pt和Ni元素仅存在于PtNi纳米合金上(图2a)。
当加热到600°C时,原始PtNi多面体结构的边角变得模糊,Zn开始从衬底渗透到纳米合金中(图2b)。在700~800℃时,这一趋势更加明显,Ni原子从金属纳米合金扩散到载体上(图2c)。Ni和Zn在900℃完成原子置换。在原子置换过程中,Zn原子从衬底富集到纳米合金颗粒,Ni原子从纳米合金迁移到CN载体,Pt原子自始至终分布在纳米合金颗粒上(图2a−d)。
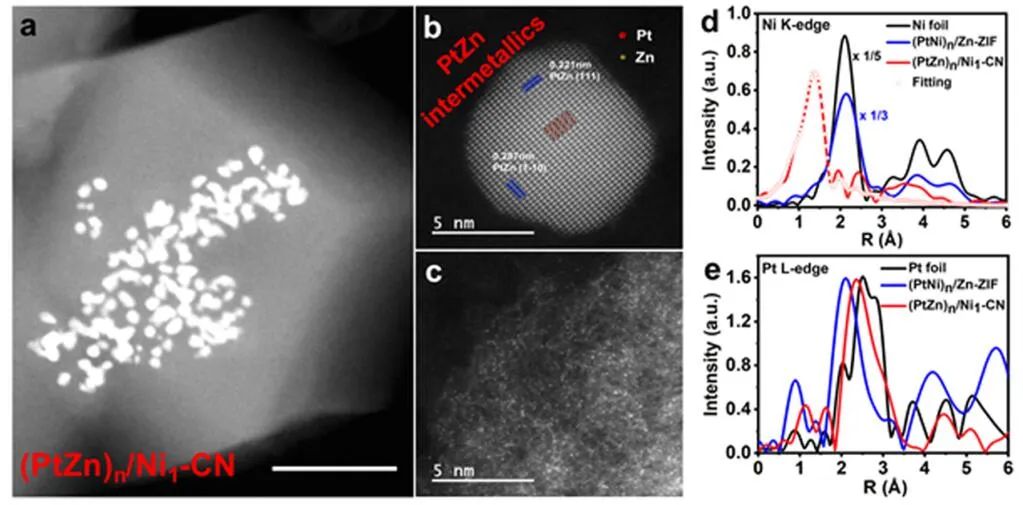
图3 (PtZn)n/Ni1-CN的结构表征。(a)(PtZn)n/Ni1-CN的HAADF-STEM图像;(b, c)单个PtZn纳米晶体和(b)CN衬底上原子分散的金属原子的AC-STEM图像(c);(PtNi)n/Zn-ZIF和(PtZn)n/Ni1-CN中Ni K边(d)和Pt L边(e)的FT-EXAFS谱。
作者对900℃热解后的产物(PtZn)n/Ni1-CN进行了表征。HAADF-STEM和TEM图像显示CN衬底发生收缩,纳米合金内部尺寸减小(图3a)。从像差校正扫描透射电镜(AC-STEM)图像分析,纳米晶相具有清晰的晶格条纹,晶格间距分别为0.221和0.287 nm(图3b),分别对应于金属间化合物PtZn的(111)和(1-10)平面,表明Zn掺杂到纳米合金相中,生成了PtZn金属间化合物。
(PtNi)n/Zn-ZIF在Ni K边的傅里叶变换EXAFS(FT-EXAFS)曲线显示了一个约2.21 Å的峰,来自Ni−M(M=Ni或Pt)散射,而(PtZn)n/Ni1-CN样品中原子分散的Ni只有一个约1.44 Å的峰,归因于Ni−N/C散射,验证了原子替换后原子分散的Ni原子的存在(图3d)。在Pt L边的FT-EXAFS曲线(图3e)中,(PtNi)n/Zn-ZIF的最近配位峰出现在2.12 Å处。Zn原子取代Ni原子后,得到的(PtZn)n/Ni1-CN由于Pt-Zn键长比Pt-Ni更长,其R空间位于2.33 Å,而(PtNi)n/Zn-ZIF的R空间位于2.12 Å。
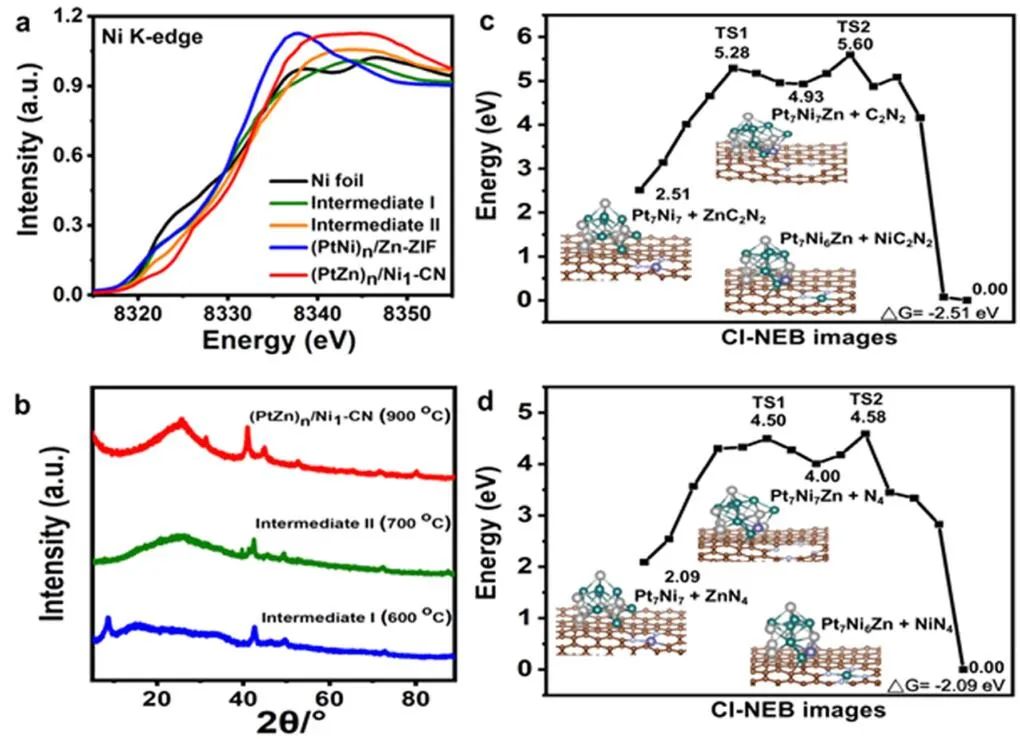
图4 (a,b)不同热解温度下,样品Ni k边的XANES(a)和XRD谱图(b);(c,d)采用CI-NEB方法计算路径上的能量分布,以及不同配位环境下相应的初始态、最终态和过渡态构型。
X射线吸收近边结构谱(XANES)表明,热解过程中,(PtNi)n/Zn-ZIF中Ni的价态随着原子置换的进行逐渐增加(图4a),表明PtNi的解构和Ni1-CN的形成。热解后,(PtNi)n/Zn-ZIF的XRD谱图没有显示Ni团簇或纳米颗粒的峰(图4b)。 作者通过密度泛函理论(DFT)计算,研究原子替换的机理(图4c,d)。从EXAFS(图3d)可以看出,(PtZn)n/Ni1-CN中Ni单原子为四配位,因此分别建立了ZnCxN4−x和Pt7Ni7模型来模拟Zn单原子和PtNi纳米合金。
从Pt7Ni7+ZnC2N2模型到Pt7Ni6Zn1+NiC2N2的交换能为−2.51 eV;从Pt7Ni7+ZnN4模型到Pt7Ni6Zn1+NiN4的交换能为−2.09 eV。原子交换过程中,经历两个阶段:第一步,Zn从ZnCxN4−x转移到PtNi合金中;第二步,Ni与CxN4−x结合。C2N2缺陷与Ni具有较大的结合能,在高温下克服反应能垒时,原子取代产物应以NiC2N2为主,而不是Ni-N4。
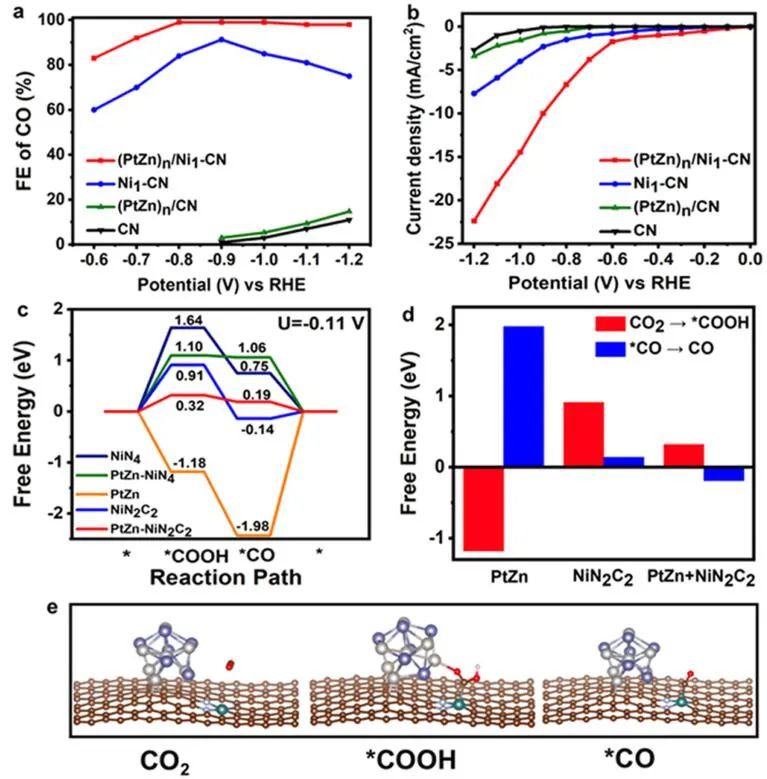
图5 不同催化剂对CO2RR的催化性能和密度泛函理论计算。(PtZn)n/Ni1-CN、(PtZn)n/CN、Ni1-CN和CN催化剂上CO电还原反应的法拉第效率(a)和不同电位下的LSV曲线(b);(c)CO2RR过程中的自由能变化;(d)CO2RR中关键步骤的自由能比较;(e)(PtZn)n/Ni1-CN催化CO2RR过程中多位点协同模型的中间结构优化。
当CO2电还原为CO时,合成的(PtZn)n/Ni1-CN与Ni1-CN、PtNi纳米合金和(PtZn)n/CN相比表现出显著增强的性能。(PtZn)n/CN和CN在电位范围为-0.9至-1.2 V时,CO的法拉第效率(FE)小于25%。单原子催化剂Ni1-CN在−0.9 V下FE可达90%。但随着工作电压的增加,FE急剧下降。
相比之下,(PtZn)n/Ni1-CN多位点催化剂在宽的电化学窗口(-0.7到-1.2 V)内表现出更高的CO选择性(>90%)。如线性扫描伏安(LSV)曲线所示(图5b),CN在−1.0 V时提供的电流密度仅为0.5 mA cm-2。在此电压下,(PtZn)n/Ni1-CN催化剂的电流密度为14.5 mA cm-2,远高于Ni1-CN(4.0 mA cm-2)和(PtZn)n/CN(1.6 mA cm-2)样品的催化活性之和,说明(PtZn)n/Ni1-CN中PtZn纳米颗粒与Ni1-CN具有协同作用。
随着电压的增加,(PtZn)n/Ni1CN的电流密度急剧增加,远远超过Ni1-CN和(PtZn)n/CN。 如图5c所示,四配位结构的Ni单原子位点形成*COOH中间体需要1.64 eV。当配位环境中的部分N替换为C时,*COOH在NiN4位点上的吸附能降低至0.91 eV,但在中等电位下仍难以达到。引入的PtZn纳米晶体通过Pt-O相互作用对*COOH中间体起到了吸附和稳定作用,PtZn-NiN2C2的质子化能量从0.91 eV显著降低到0.32 eV。
但PtZn-NiN4的质子化能量仍然较高,这意味着*COOH中间体更容易在PtZn-NiN2C2位点上形成。另一方面,*CO的脱附对CO的生成也很重要。从热力学角度看,尽管CO2在PtZn纳米晶体上的直接质子化过程在能量上更有利,约为−1.18 eV,但最后一步对*CO的脱附而言非常困难。因此,与单个PtZn或Ni原子相比,多位点PtZn-NiN2C2可以同时促进质子化和*CO脱附(图5d)。
总结与展望
本工作发现了单原子与纳米合金之间发生原子置换的新现象,丰富了纳米粒子与单原子之间的转化策略。原子置换也为合成多活性位点催化剂建立了新的策略。DFT计算揭示了原子置换机理,并证明了制备的多活性位点催化剂具有协同催化作用。
审核编辑:刘清
-
快速锁定高频准的原子钟 #原子钟厂家 #芯片原子钟 #微型铷原子钟 #同步天下 #铷jf_16650182 2026-04-03
-
使用MACMode原子力显微镜处理液体中的金纳米粒子应用笔记2019-10-23 1916
-
什么是原子电池?2009-11-03 2002
-
基于FPGA的单原子反馈控制与LabVIEW和NI FlexRIO的介绍2017-10-15 779
-
纳米级原子力显微镜diy图解2018-09-20 12405
-
科学家捕获到单个原子观察到了原子间相互作用2020-02-24 3045
-
原子级工艺实现纳米级图形结构的要求2020-06-02 3360
-
简述石墨烯纳米结构的原子级精准构造2021-06-17 5034
-
界面化学键原子水平调控异质结光催化剂性能!2022-08-18 2298
-
Adv Mater综述:非贵金属单原子/双原子在光合成的应用2023-05-15 2103
-
中科院大连化物所《Angew》:开发出单原子合金材料2023-07-05 1245
-
双原子催化剂综述:适用于能源和环境催化的双原子催化剂2023-07-17 10512
-
如何使用原子类型2023-11-10 1939
-
什么是原子层刻蚀2025-01-20 1828
-
铷原子钟与CPT原子钟有哪些区别呢2026-02-12 7120
全部0条评论

快来发表一下你的评论吧 !
