

付超鹏Angew:聚合物基p型正极实现超长循环铝电池
描述
研究背景
铝离子电池具有储量丰富、成本低、安全性好、理论容量高等优点,是一种很有前景的大规模储能技术。但是,具有无机正极的铝离子电池在循环过程中仍存在动力学缓慢、结构坍塌等问题。
成果简介
近日,上海交通大学付超鹏副教授在Angew上发表了题为“Phenoxazine Polymer-based p-type Positive Electrode for Aluminum-ion Batteries with Ultra-long Cycle Life”的论文,提出了一种p型聚(乙烯基苄基-N-吩恶嗪)(PVBPX)正极用于AIBs。双活性位点使PVBPX能够在0.2 A g−1下提供133 mAh g−1的高容量。PVBPX的扩展π共轭结构、不溶性和无键重排的阴离子氧化还原化学实现了50000圈的超长循环寿命。实验和理论结果均证明,AlCl4−离子能够可逆地与PVBPX中的N位和O位配位/解离,从而实现电荷存储。
研究亮点
(1)本文通过对AIBs的一步亲核取代反应,设计并制备了p型正极材料聚乙烯基苄基-N-吩恶嗪(PVBPX)。
(2)PVBPX中的N和O双活性位点实现了133 mAh g−1的高比容量。此外,接枝在聚合物上的双活性位点可以显著抑制聚合物在电解质中的溶解。
(3)PVBPX的不溶性,扩展的π共轭体系以及无键重排的阴离子氧化还原化学加快了反应动力学,并实现了超长的循环寿命。
图文导读
聚合物PVBPX的合成过程如图1a所示,通过一步亲核取代反应将吩恶嗪(PX)单体与线性聚(乙烯基苄基氯)接枝,以获得PVBPX。图1b的傅里叶变换红外(FTIR)光谱证实,吩嗪单元成功接枝到聚合物支链上。吩恶嗪的特征吸收峰在1700至700cm−1范围内。PX中3390 cm−1处的N-H基团特征峰在PVBPX中消失,表明亲核取代后氨基上的氢原子被线性聚合物的苄基取代。结果表明,PX单体成功接枝到聚合物支链上。
PX单体显示出高结晶度结构,然而,制备的PVBPX没有显示出明显的衍射峰,并显示出无定形结构(图1c)。PX单体在约150°C下分解和升华,并在270°C下失去所有重量。然而,PVBPX在280°C的更高温度下分解,且PVBPX聚合物在270°C时的重量损失小于2.5%,显示出优异的热稳定性(图1d)。
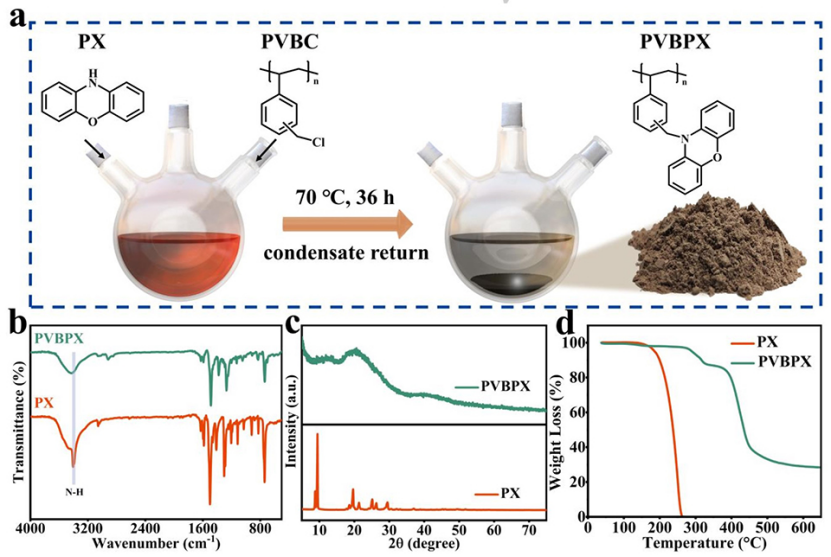
图 1、PVBPX聚合物的(a)合成示意图。(b)FT-IR光谱。(c)XRD图谱。(d)PVBPX和PX在N2气氛中的TG曲线。
以AlCl3/[EMIm]Cl离子液体电解质,PVBPX正极与Al负极组装了AIBS,如图2a所示。PVBPX正极的循环伏安法(CV)曲线在1.07/1.0V、1.81/1.72V附近出现两对氧化还原峰(图2b),表明PVBPX中活性位点发生了可逆的氧化还原过程。PX正极的CV曲线峰电位与PVBPX峰电位相似,说明PVBPX聚合物的氧化还原过程发生在吩恶嗪单元内。两对氧化还原峰(O1/R1和O2/R2)可能分别归因于-N和-O-活性位点上的氧化还原反应。带有PX和PVBPX的AIBs恒流充放电曲线在1.0V和1.7V显示出两个不同的放电电压平台(图2c)。
在0.2 A g−1下,PVPBX电极放电容量为133 mAh g−1,首圈库仑效率仅为83%,这与固体电解质界面的形成有关。图2d显示,PVBPX正极表现出优异的倍率性能。此外,即使在高电流密度下,PVBPX的充放电电压平台也保持良好(图2e)。在200 mA g−1下,PVBPX正极循环100次后没有容量衰减,而PX正极在100次循环之后,仅保持85%的容量(图2f)。图2g显示,PVBPX正极在50000次循环后保持88 mAh g−1的比容量,每个循环没有明显的容量衰减,在50000次后库仑效率保持99.8%。图2h显示,在循环50000次后,FTIR光谱中仍然检测到C-N(1375和1265 cm−1)和C-O(1127 cm−1)的吸收峰,表明PVBPX的分子结构在长循环后保持完整。
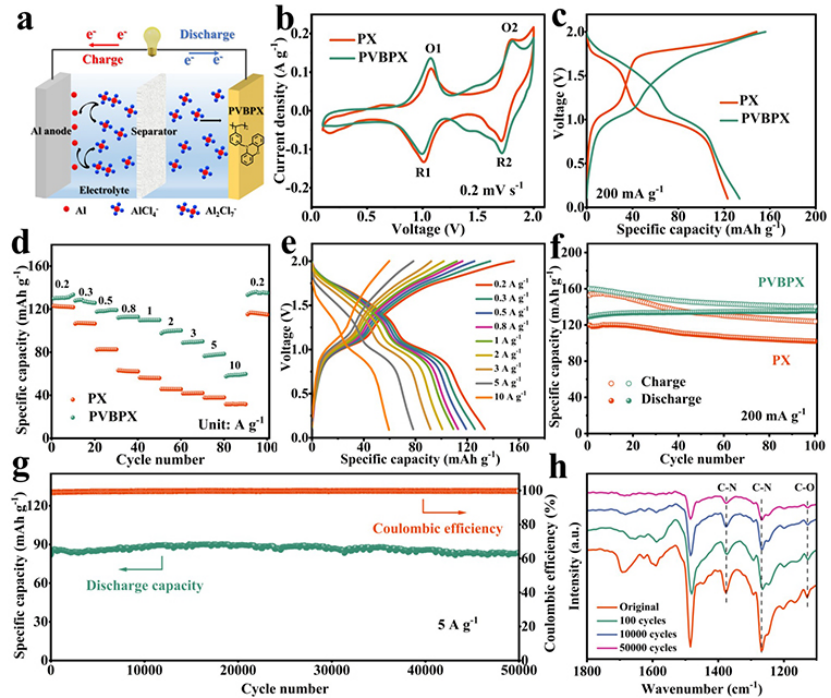
图 2、(a)带有PVBPX正极的AIBs示意图。(b)具有PVBPX和PX的AIBs在0.2 mV s−1扫速下的CV曲线。(c)0.2 A g−1下的充放电曲线。(d)倍率性能和(e)各种电流密度下的充放电曲线。(f)PVBPX和PX在0.2A g−1下的循环性能。(g)PVBPX和PX在5 A g−1下的长循环稳定性。(h)不同循环圈数下PVBPX电极的FTIR光谱。
图3a中不同扫速下的CV曲线显示,峰电流随着扫速的增加而增加,峰值电流(i)和扫速(v)之间的关系可以写为:
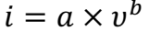
其中a和b为可调参数。b值可由log(i)与log(v)图的斜率确定。如果b值为0.5,则表明充放电过程由离子扩散控制。如果b值为1.0,则电化学行为受表面电容支配。根据图3b中的log(i)对log(v)图,与四个峰O1、O2、R1和R2对应的b值分别为0.849、0.765、0.895和0.904。结果表明,PVBPX的电化学过程同时受到离子扩散和电容的影响。此外,表面电容贡献和离子扩散贡献的比率可以通过以下方程进一步计算。
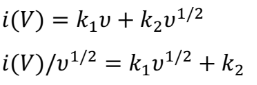
其中k1v和k2v1/2分别对应于表面电容和离子扩散贡献。图3c显示,随着扫速从0.2增加到5 mV s−1,PVBPX的表面电容贡献从63.7%增加到89.1%,高于相应扫速下的PX电极(图3d)。计算表明,PVBPX正极的充放电过程主要由表面赝电容贡献,这促进了快速的电荷存储,实现了出色的倍率性能。
图3e的电化学阻抗谱(EIS)显示,与PX电极相比,PVBPX电极在高频区域显示出更小的直径,在低频区域显示出更陡的斜率,表明PVBPX电极具有小的电荷转移电阻(Rct)和低的离子扩散电阻。PX和PVBPX电极的Rct在20次循环后都显著降低,较小的Rct反映了高的电导率和快速的反应动力学。
图3f显示,PVBPX正极不仅提供了最佳的循环稳定性和倍率性能,还表现出具有竞争力的能量密度和放电电压。因此,PVBPX是一种很有前途的可充电AIBs正极材料。
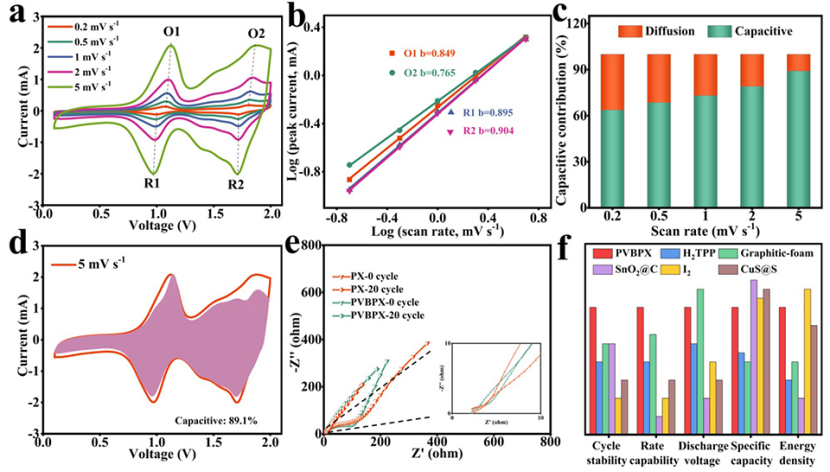
图 3、(a)在0.2至5 mV s−1不同扫速下的CV曲线。(b)氧化还原峰处log(v)和log(i)的线性关系。(c)不同扫速下的电容贡献。(d)5 mV s−1下的赝电容贡献。(e)循环之前和20个循环之后的奈奎斯特图。(f)AIBs中不同正极材料的关键电化学参数比较。
原位XPS光谱显示,N 1s光谱在399.4 eV处显示出单峰(图4b)。充电至1.4V后,峰向更高的结合能移动,在N 1s光谱中401.4eV处出现新峰,表明C-N-C部分氧化为带正电的C-N+-C。氧化的N+部分占总N原子的71.4%。充电至2.0V后,C-N+-C的分数达到72.5%。与充电至1.4V相比,氧化C-N+-C的分数没有显著差异,表明作为活性位点的氮原子在充电过程中被氧化,反应主要发生在1.4 V以下。
在O 1s光谱中可以观察到类似的现象(图4c),其中对应于氧化态的新峰出现在充电态的高结合能处。在充电状态下,电荷离域导致N和O核的电子密度降低,这导致相应的XPS光谱向更高的结合能移动。与N 1s光谱不同,充电至1.4 V时,氧化峰(C-O+-C)的强度在O 1s光谱处较弱。然而,充电至2.0 V时,C-O+-C的峰明显增强,表明O位的氧化电位高于N位。在随后的放电过程中,氧化的C-N+-C和C-O+-C峰可逆地减弱,表明N和O活性位点表现出高的氧化还原可逆性。结合N 1s和O 1s光谱的分析,在~1.0 V的电压平台是N位的氧化还原反应,而在~1.7 V的电压平台则是O位的氧化反应。此外,Al 2p光谱的峰强随着充电而逐渐增加,并在随后的放电过程中可逆地减弱(图4d)。虽然在Cl 2p XPS光谱中观察到类似的趋势,但充放电过程分别伴随着Cl信号的增强和减弱(图4e)。这一结果表明,在充放电过程中,Al和Cl都参与了配位/解离反应。
图4f的原位EPR显示,在充电状态下观察到的g值为2.005,表明在充电过程中阳离子自由基的形成。同时,该结果证实阳离子PVBPX与带负电的载体配合,进行阴离子氧化还原反应。在随后的放电过程中,阳离子自由基的信号可逆地消失,进一步证实PVBPX的氧化还原过程是高度可逆的。
图4g显示,PVBPX正极的电荷存储经历两步可逆过程。在充电过程中,N位首先被氧化成带正电的状态,同时与AlCl4-载流子结合。随后,O位点被氧化形成自由基阳离子,该自由基阳离子也与AlCl4-发生配位反应。在放电过程中,阴离子的脱配位反应同样是伴随着氧化态逐渐还原的两步反应过程。
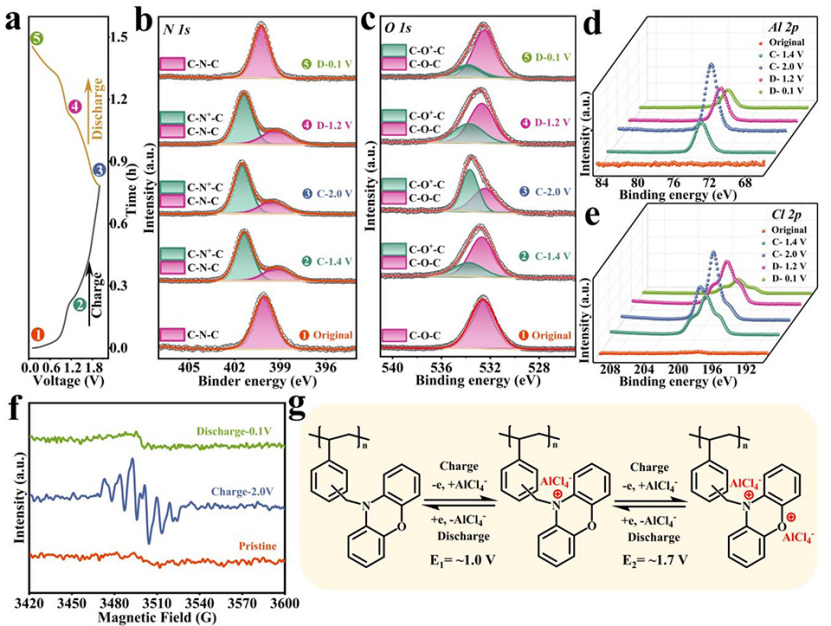
图 4、(a)200 mA g−1下PVBPX正极的充放电曲线。(b)PVBPX正极在不同电压下的N 1s、(c)O 1s、(d)Al 2p和(e)Cl 2p XPS光谱。(f)PVBPX电极的原位EPR谱。(g)PVBPX的电化学氧化还原机理。
中性PVBPX、单电子氧化产物(PVBPX+)和双电子氧化产物(PVBPX2+)的分子静电势(ESP)分布图,如图5a所示。具有正静电势的蓝色区域倾向于亲核位点,并且容易参与亲核反应。具有负静电势的红色区域可以充当亲电子位点,并倾向于进行亲电子反应。在中性PVBPX的ESP图中,杂原子两侧的两个苯环区域表现出明显的负电荷特征,具有强烈的给电子倾向。而氧化PVBPX+和PVPBX2+的ESP图在整个分子中显示出强烈的正电荷特性,特别是在中间杂环的N和O区域。ESP图谱的计算结果从理论上证明,N和O是PVBPX分子中亲核反应的活性位点,并与电解质中带负电的载体配位。
此外,通过诱导电流密度(AICD)的各向异性研究了PVBPX及其氧化产物的芳香性变化。如图5b所示,中性PVBPX分子的N-O杂环两侧的苯环表现出强烈的芳香特征,而中心N-O杂环表现出显著的反芳香特征。在PVBPX分子失去一个电子后,N-O杂环从反芳香特性转变为非芳香特性。失去另一个电子后,PVBPX2+的AICD图呈现顺时针循环,N-O杂环转变为芳香性。这种反芳香族转化遵循Huckel法则,在PVBPX的两个氧化还原过程中只有π电子离域,这表明氧化中间体PVBPX+和PVBPX2+都是热力学稳定的。
基于DFT计算了最低未占分子轨道(LUMO)和最高已占分子轨道的能级(HOMO)(图5c)。中性PVBPX的前沿分子轨道居群及其氧化态表明,只有π-电子参与氧化还原反应过程,没有观察到键重排,证实了PVBPX在氧化还原反应期间的结构稳定性。此外,与中性PVBPX相比,PVBPX+和PVBPX2+的能隙明显更低,这有助于充电期间PVBPX与阴离子的相互作用,促进电子转移,并减少循环期间的极化。
此外,通过DFT优化了PVBPX分子及其与AlCl4−配位的配合物构型(图5d)。在充电过程中,一个AlCl4−首先与PVBPX中的N原子配位,然后位于远离乙基侧链的一侧。随着进一步充电,第二个AlCl4−与O原子相互作用,然后位于杂环平面的另一侧,使总能量最小化。放电过程是充电过程的可逆反应。
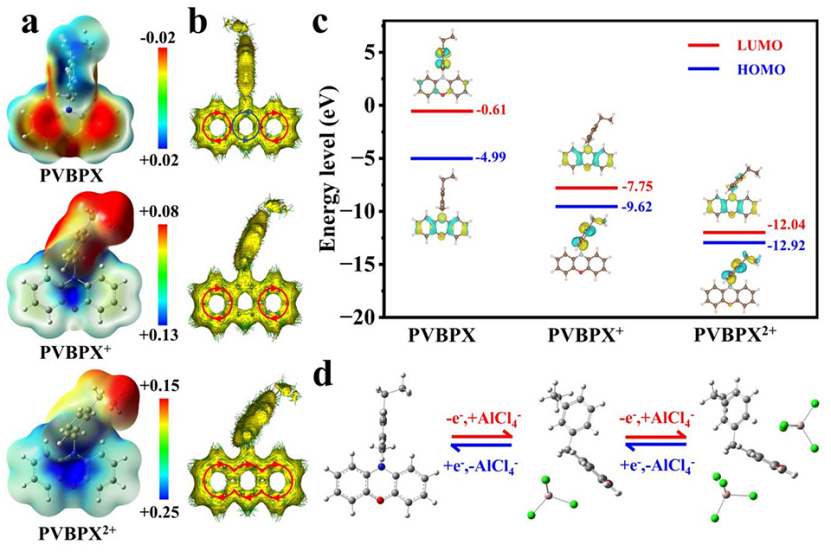
图 5、PVBPX及其氧化物种(PVBPX+和PVBPX2+)的(a)分子ESP图。(b)ACID图。(c)LUMO-HOMO能级。(d)PVBPX电极的两步氧化还原过程和分子几何结构。
总结与展望
本工作成功开发了用于高性能AIBs的p型有机正极PVBPX。PVBPX中的双活性位点有助于实现高比容量,设计的结构和独特的配位反应保证了超长的循环寿命。各种表征技术和DFT计算表明,N和O是PVBPX中的双氧化还原中心,在0.2 A g−1下有133 mAh g−1的高比容量,扩展的π-共轭结构和无键重排的可逆阴离子氧化还原化学使得PVBPX能够循环长达50000次。因此,p型有机聚合物是用于可充电AIBs的一种有前途的电极材料。
审核编辑 :李倩
-
复旦大学王永刚Angew:可降解的自由基聚合物锂电池正极材料2025-01-02 1447
-
锂聚合物电池技术参数_锂聚合物电池构造2020-08-03 15122
-
锂聚合物电池的原理是什么?2020-03-13 2410
-
锂电池VS聚合物锂电池,谁才是未来的主角?2018-08-17 7282
-
聚合物锂电池和锂电池区别是什么?2017-09-14 5106
-
聚合物锂电池的倍率用什么代表?2015-11-07 3987
-
锂聚合物电池2013-07-09 2878
-
聚合物锂电池工作原理表现2013-06-14 3944
-
聚合物锂离子电池的构成2013-06-06 3471
-
聚合物锂电池的生产2013-05-10 4088
-
对聚合物锂电池的优点和缺点进行分析2012-11-29 5150
-
聚合物锂电池充电电压简述2011-04-18 3837
-
什么是聚合物电池2009-05-24 7730
全部0条评论

快来发表一下你的评论吧 !
