

阐明采用不同电解质的水系Zn/MnO2电池的反应机理
描述
研究背景
由于安全性和低成本,可逆水系Zn/MnO2电池(AZMOBs)成为一种有前途的电网级储能替代方案。
近年来,由于需要充分了解其电荷存储机制,以提高其效率和循环寿命,采用弱酸电解质的AZMOBs引起了人们的广泛关注。
不同弱酸性电解质的AZMOBs中Mn的氧化还原机理目前仍存在争议,主要有以下几种:Zn2+/H+嵌入机制,Zn2+/H+嵌入诱导MnO2-ZnxMnOy/MnOOH转换机制和Mnn+溶解−沉积机制。
最近,使用原位同步加速器X射线荧光谱(XFM)提供了MnO2溶解-沉积是ZnSO4电解质中主要的Mn氧化还原反应的直接实验证据。然而,XFM方法无法提供结构信息,也无法提供固体正极或电解质内配位环境的演变信息。
成果简介
鉴于此,石溪大学的Kenneth J. Takeuchi(通讯作者)等人利用Mn K边XAS技术,对ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2水溶液的锌电池的α-MnO2溶解-沉积氧化还原过程进行了实验研究,此项技术能够同时表征参与Mn氧化还原反应的液相和固相,最后阐明了采用不同电解质的水系Zn/MnO2电池的反应机理。
研究亮点
1、不同电解质(ZnSO4,Zn(CF3SO3)2和Zn(CH3COO)2)中,MnO2具有相似的锰配位环境,但固相和液相中Mnn+物种的定量分布存在差异;
2、拉曼光谱证明,在正极处,电荷作用下形成了结晶性差的含锰产物,并采用TEM对沉积物的形态和表面状况进行了深入研究。
图文介绍
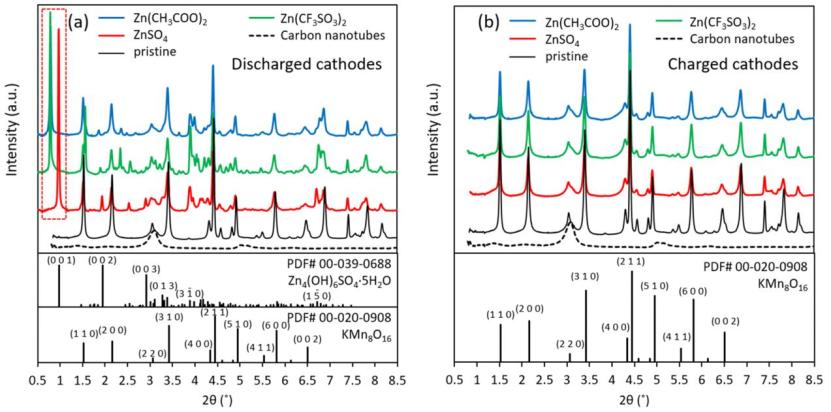
图1 在ZnSO4、Zn(CF3SO3)2或Zn(CH3COO)2电解质中循环的(a)放电和(b)带电正极与原始电极和碳纳米管的同步X射线衍射图。
采用基于同步辐射的XRD技术对不同电解质中循环至第一次放电和第一次充电的α-MnO2正极进行了表征(图1)。
放电时,所有正极中都出现低于1°的强峰(图1a红色虚线框),表明形成了新相。以往文献表明,ZnSO4,Zn(CF3SO3)2和Zn(CH3COO)2电解质中形成的新相分别为Zn4SO4(OH)6·xH2O(ZHS),Zn5(OH)8(CF3SO3)2·xH2O,(ZHT)和Zn5(OH)8(CH3COO)2·xH2O(ZHA)。
充电后正极的XRD图谱中,ZHS、ZHA和ZHT峰消失,只留下原始材料的峰(图1b),为未反应的α-MnO2。α-MnO2峰值强度较原始状态电极的低,表明其质量分数降低。

图2 在(a,d)ZnSO4电解质、(b,e)Zn(CF3SO3)2电解质和(c,f)Zn(CH3COO)2电解质中放电的α-MnO2正极的透射电镜表征显示了α-MnO2棒和沉积的板状材料的图像(a-c)和板状材料的衍射图(d-f)。
在电池初次放电(图2)和初次充电(图3)后,采用TEM观测了α-MnO2正极。在首次放电时,三种电解质中的α-MnO2纳米棒在表面和棒的末端附近都显示出溶解现象(图2a−c),在纳米棒旁边还观察到板状沉淀物质。ZnSO4电解液中放电产物的衍射模式与已知结构羟基硫酸锌(ZHS)匹配性很好(图2d)。

图3 (a)ZnSO4,(b)Zn(CF3SO3)2和(c)Zn(CH3COO)2电解质中α-MnO2正极在首次充电循环后的TEM图像显示了充电过程中表面沉积的形成;(d-f)不同电解质下的嵌入功率谱。
ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2电解质中,第一个带电电极的成像如图3所示。三种电极类型的样品均含有棒状α-MnO2,电极表面镀有电化学沉积物质,沉积的物质可能由含Zn/Mn相组成。
在图3d-f中,嵌入功率谱中的红色圈出的信号对应于Zn/Mn相。且过滤HRTEM图像表明,表面沉积物都优先排列在α-MnO2棒上。
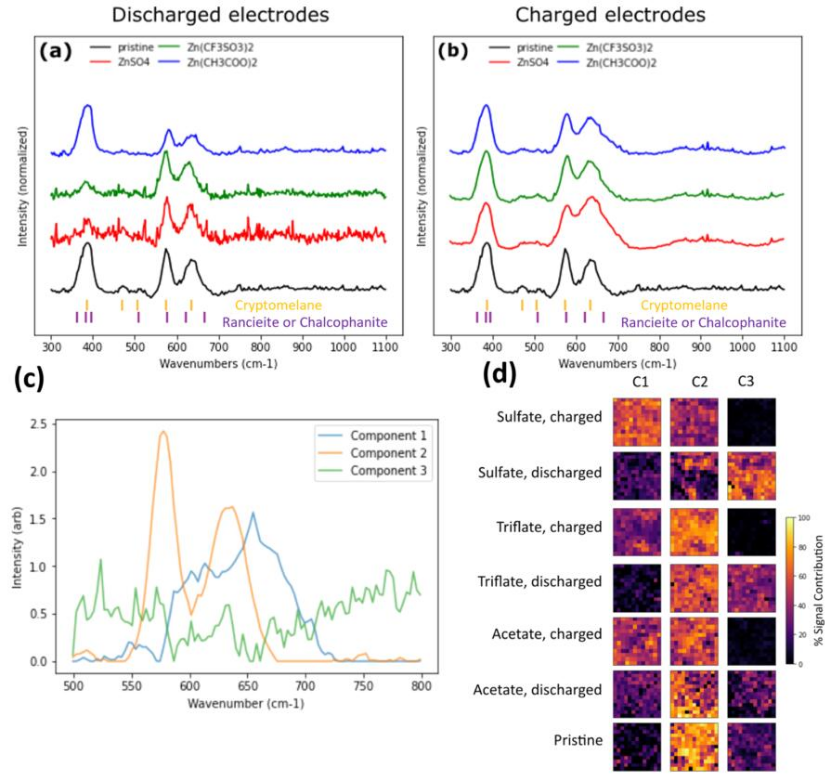
图4 (a)放电电极和(b)带电电极的拉曼光谱;(c)未混合共聚焦拉曼数据集的NMF分量。对提取的信号组分进行识别,其中组分1为层状相,组分2为锰钾矿,组分3为残留背景;(d)非原位电极表面已识别组分的空间分布图。
从原始电极上收集的拉曼光谱显示,仅有与α-MnO2相关的峰,出现在387、471、494、507、577和635 cm−1。
在ZnSO4、Zn(CH3COO)2和Zn(CF3SO3)2电解质中放电电极的拉曼光谱与原始电极和α-MnO2材料相似(图4a),表明放电电极中存在未反应的残余α-MnO2。
充电后,在667 cm−1附近出现了一个新峰,表明形成了层状Zn−Mn−O相(图4b)。
对拉曼数据集进行NMF分量,其中组分1为层状相,类似于层状钙硬锰石(Ca,Mn2+)0.2(Mn4+,Mn3+)O2·0.6H2O或黑锌锰矿ZnMn34+O7·3H2O。
通过比较不同电解质和充放电条件下层状相(成分1)的NMF归一化信号强度,可以观察到层状相仅在充电电极中存在,这表明在充电过程中形成层状相,在放电过程中消失(图4d)。
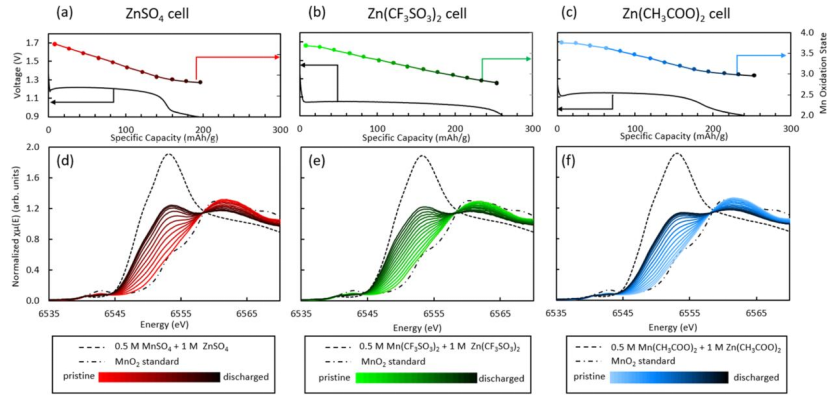
图5 使用LCF计算的(a)ZnSO4,(b)Zn(CF3SO3)2,(c)Zn(CH3COO)2的操作单元放电期间的平均Mn氧化态的电压分布;(d)ZnSO4,(e)Zn(CF3SO3)2和(f)Zn(CH3COO)2电池的原位XANES演化。
对每次XANES扫描进行线性组合拟合(LCF),利用KMn8O16(s)值和相应的溶液Mn2+标准,获得电池的平均Mn氧化态(OS)。
在放电期间,XANES的X射线吸收边缘转移到较低的能量处(图5d−f),这种变化表明平均Mn氧化态的降低,与由LCF确定的Mn平均氧化态变化一致(图5a−c)。
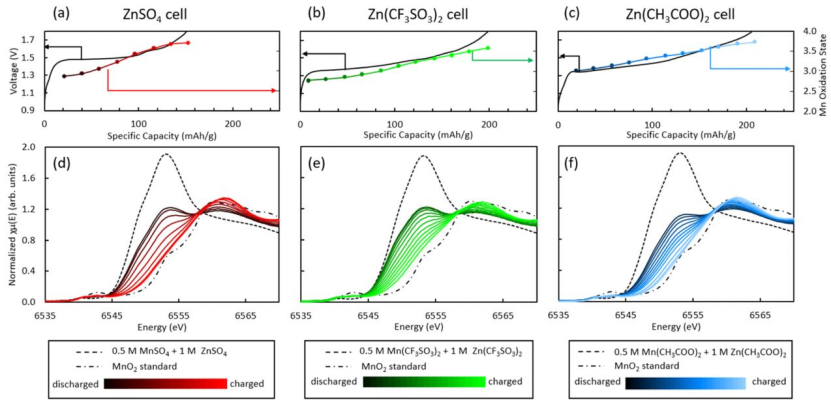
图6 使用LCF计算的(a)ZnSO4,(b)Zn(CF3SO3)2,(c)Zn(CH3COO)2的操作单元充电期间的平均Mn氧化态的电压分布。充电时,(d)ZnSO4,(e)Zn(CF3SO3)2和(f) Zn(CH3COO)2电池的原位XANES演化。
ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2电池在充满电后对应的平均Mn氧化态值分别略低于原始材料值3.73、3.69和3.75。
这种氧化态变化归因于充电产物与原始材料之间的结构和化学差异。如拉曼光谱表明,充电产物是层状的锌锰氧化物,这种材料通常由MnOx层组成,层间插入Zn2+离子。
与含有一价K+的原始α-MnO2相比,由于Zn2+的存在,使得放电产物具有较低的氧化态。
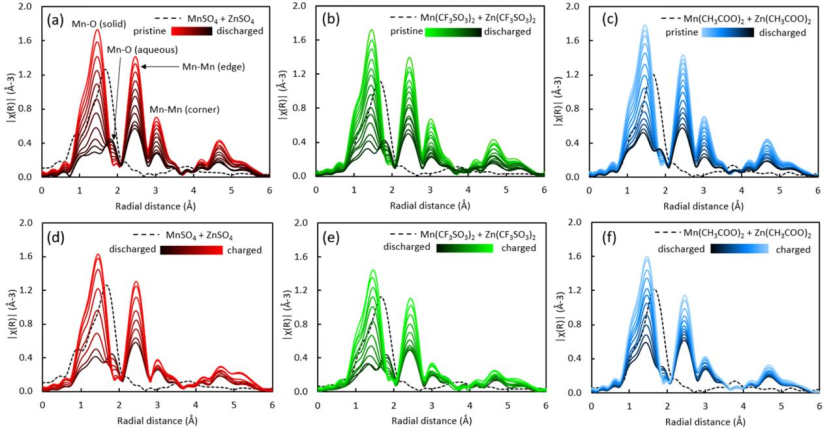
图7 (a)ZnSO4,(b)Zn(CF3SO3)2和(c)Zn(CH3COO)2电池放电期间,原位EXAFS在r空间的演化;(d)ZnSO4,(e)Zn(CF3SO3)2和(f)Zn(CH3COO)2电池在充电过程中相应的EXAFS演化。
将原位EXAFS光谱傅里叶变换到径向空间(r-空间),在r-空间中可以区分配位环境的变化。图7显示了在r-空间中初始放电和后续充电期间三个电池的原位EXAFS演化。
三种电池的r-空间EXAFS光谱具有相似的主峰。如图7a所示,在∼1.5 Å处的第一个主峰对应固体MnO2结构内的第一层Mn−O散射路径,在∼2.5 Å处的第二个主峰对应第二层Mn−Mn散射路径,在∼3.0 Å处的第三个主峰对应第三层Mn−Mn散射路径。
第二层Mn-Mn散射路径代表了MnO2结构中两个共享边的MnO6八面体的相对位置,而第三层Mn-Mn散射路径代表了α-MnO2结构中两个共享角的MnO6八面体的相对位置。
三个电池EXAFS光谱在初始放电过程中表现出相似的形状,而始放电过程中,r空间峰值强度逐渐降低,并存在~1.9 Å处新峰的增长。与EXAFS标准Mn2+水溶液的对比表明,该峰可能对应于溶剂化[Mn(H2O)6]2+离子的第一层Mn−O散射路径。
在充电时,除了第三层Mn-Mn峰外,所有的Mn-Mn峰都恢复到原始强度,三个电池的水Mn-O峰都消失了。
充电的α-MnO2与原始的α-MnO2在EXAFS光谱上的差异表明,在充电时,形成了一个具有不同于原始α-MnO2的Mn中心局部结构的产物。
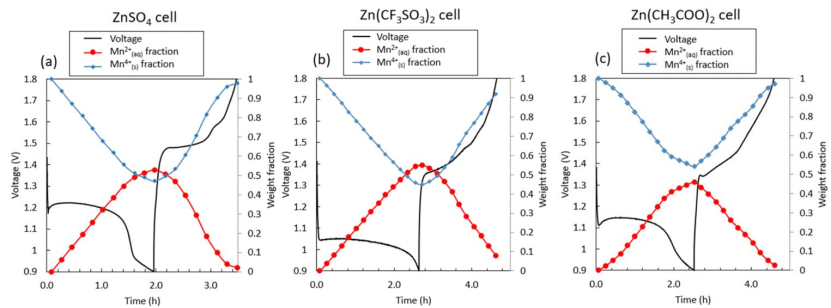
图8 XANES-LCF结果显示(a)ZnSO4,(b)Zn(CF3SO3)2和(c)Zn(CH3COO)2电池在第一个循环中的溶液中/固相中Mn的质量分数。
Mn的溶解-沉积过程包括溶解的溶剂化Mn2+离子和固体MnOx。使用标准Mn2+水溶液,对相应的电池进行原始扫描,以获得电化学还原和氧化过程中单元内的水和固体Mn质量分数(图8)。
在三种电池的初始放电过程中收集的XAS光谱表明,LCF拟合的固体/水Mn的重量分数呈现与电化学过程相关的Mn溶解现象(图8)。
在初始放电结束时,约50%的Mn从正极溶解形成Mn2+。在充电过程中,虽然大多数溶解的Mn2+以固体形式重新沉积,但少量的Mn2+仍未被氧化。
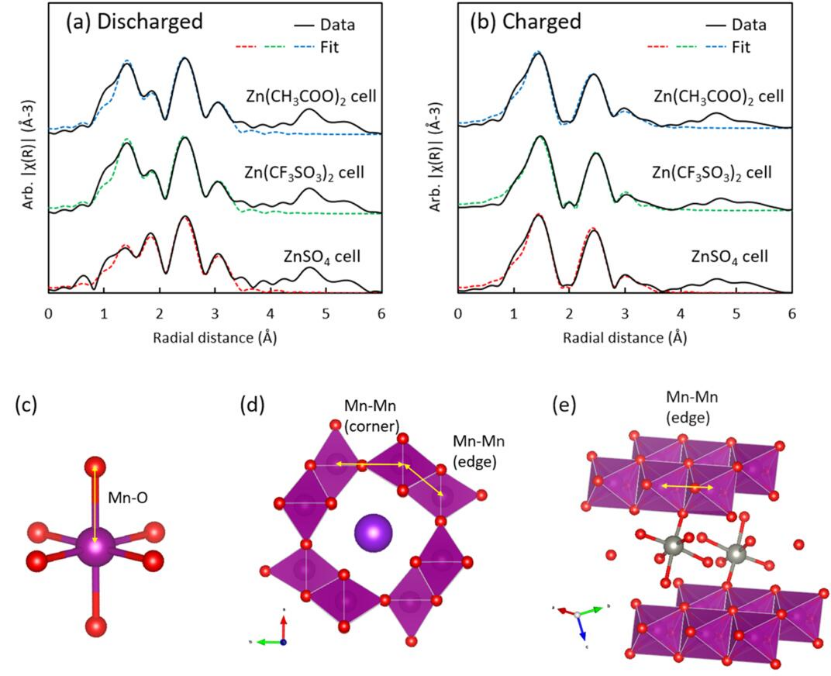
图9 (a)完全放电和(b)充满电的电池扫描的EXAFS拟合结果;(c−e)EXAFS拟合时用于执行FEFF计算的理论结构。
采用EXAFS拟合方法,利用理论FEFF计算结构对含锰产物进行解析。[Mn(H2O)6]2+结构可以用[MnO6]理论结构很好地表示(图9c)。
初始放电时,原始α-MnO2溶解在ZnSO4、Zn(CF3SO3)2或Zn(CH3COO)2电解质中形成水合[Mn(H2O)6]2+离子,未溶解的α-MnO2没有改变其局部结构。
对于充电电池的原位EXAFS光谱,放电时出现的Mn2+峰消失了,只在前三个壳层上留下三个主要峰。除了第三层Mn-Mn峰明显减少,这些光谱与原始物相扫描光谱相似(图7)。
这表明在充电正极中,Mn的角共享[MnO6]八面体的数量显著减少。精细EXAFS拟合结果表明,在充电过程中,大部分Mn2+会转化为固态ZMO,而未反应的KMO在电池的充电过程中都没有发生结构变化。
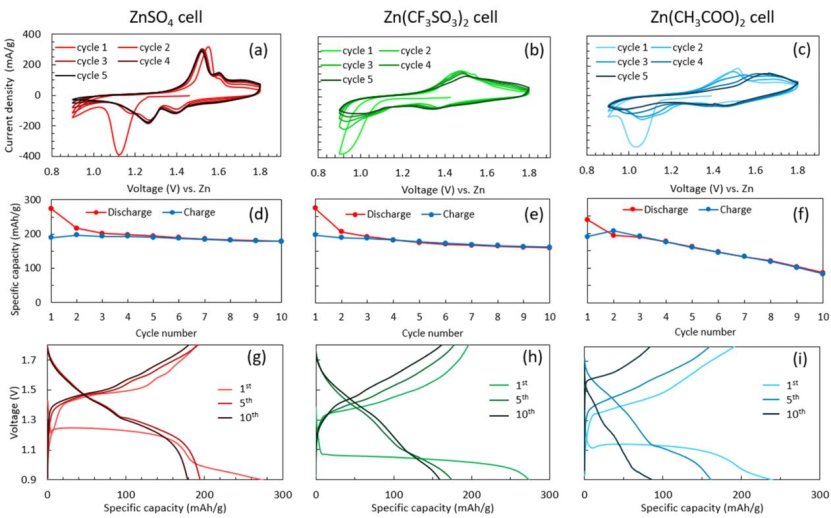
图10 (a-c)5个周期的CV结果和(d-f)10个周期的容量保持曲线;(g,h)ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2电池在10个周期内的电压分布演变。
三个电池的初始放电都以一个平坦的电压平台开始,但它们的开路电压不同,ZnSO4电池显示出最高的电压。为了证明三种电解质体系之间的电化学差异,测试了这三种体系的CV(图10a−c)和循环图(图10d−i)。放电平台的差异很可能是由于阴离子效应造成的。
CF3SO3-阴离子具有更高的静电电位,因此与SO42-阴离子(MPI=10.47 eV)相比,CF3SO3-阴离子具有更低的分子极性指数(MPI)(4.68 eV),这导致CF3SO3-阴离子具有更高的疏水性。因此,SO42−阴离子倾向于与H2O结合,在正极表面附近形成富水环境,而CF3SO3−和CH3COO−阴离子倾向于吸附在正极上,形成贫水环境。
这种贫水环境为锰溶解所需的质子化过程产生动力学障碍,降低了放电电压平台。相应地,在充电过程中,CF3SO3−或CH3COO−电池中体积较大的阴离子会减少Mn2+离子周围的H2O水分子数量,减轻溶剂化效应,从而增强电荷和离子转移,降低电荷电压平台。
对于Zn(CH3COO)2电池,在10个周期内观察到明显的容量衰退。这可能是由于CH3COO−和Zn2+离子之间的强结合,导致CH3COO−在充电时从ZHA中缓慢提取,因此限制了Mn2+插入到ZHA中,最终抑制了Mn的沉积。
总结与展望
本文采用Mn K边原位XAS技术,对ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2水溶液锌电池的α-MnO2溶解-沉积氧化还原过程进行了实验研究。
EXAFS数据的分析采用了多相方法,分析了固态和溶解的过渡金属成分。结果表明:三种弱酸性电解质的锰溶解-沉积过程具有相似的配位环境,但锰在固相和溶液中的分布不同。
原位XAS表征方法可以独立研究固相或电解质中的反应。该方法为在复杂环境中对结晶性差的多相材料的表征提供了一种实用方法。
审核编辑:刘清
-
不同类型的电池的电解质都是什么?2024-02-27 4441
-
一种新型的ZnSO4-基共晶电解质介绍2023-08-14 3958
-
MnO2钽固体电解电容器介绍2023-06-20 4042
-
水系锌离子电池电解质设计原则2023-05-30 6209
-
新型水系电解质实现长循环寿命的高压水系锂/钠离子电池2022-12-09 4517
-
设计Zn2+溶剂化结构/壳层提高锌负极容量利用率2022-11-28 3791
-
一种用于水系锌离子电池的新型碳基聚合物PODA/MnO2杂化正极材料2022-11-09 3036
-
具有高柔韧性和稳定性的Zn//CNT@MnO2柔性电池2022-07-12 3620
-
锂二氧化锰电池有什么特点?2020-03-10 2718
-
碱性Zn-NiOOH/MnO2一次电池的性能研究2011-03-10 2223
-
锂离子电池聚合物电解质导电机理2009-12-09 2863
-
锂锰电池的反应机理2009-11-06 7190
-
锂离子电池聚合物电解质导电机理是什么?2009-10-29 7848
-
电池内的电解质是什么?2009-10-20 1262
全部0条评论

快来发表一下你的评论吧 !
