

利用In2.77S4/多孔有机聚合物光催化还原CO2制乙烯
描述
【研究背景】
近年来,对化石燃料的依赖增加了大气中的CO2含量,导致全球变暖。自20世纪70年代以来,科学家们一直在努力模仿自然光合作用,以将大气中的CO2转化为碳水化合物和其他增值化学品,以减轻全球变暖。在各种可用策略中,光化学将 CO2 还原为碳氢化合物燃料和其他高价值化学品近年来引起了广泛关注。已经探索了多种复杂的多相光催化系统,例如金属氧化物、金属硫化物、沸石、多孔有机聚合物 (POP)、金属有机骨架 (MOF) 和共价有机骨架 (COF),以提高 CO2转化率。
然而,由于将CO2选择性地转化为多碳 (C2+) 产物如C2H4需要多电子转移和C-C偶联,选择性受到严重阻碍。反应过程中在催化位点产生的C1中间体也导致对C2+产物的选择性差。因此,调控催化剂表面,调控C1中间体与活性位点之间的结合能,实现高效光化学CO2-to-C2H4转化具有重要意义。
【成果简介】
印度化学技术研究所John Mondal,贾瓦哈拉尔·尼赫鲁高级科学研究中心Sebastian C. Peter教授报告了一种无模板、经济高效的合成策略,以开发基于咔唑的多孔有机聚合物 (POP) 复合催化剂。该复合催化剂由In2.77S4和多孔有机聚合物 (POP) 组成,通过静电相互作用结合在一起。利用催化活性In中心和光捕获POP的协同作用,该催化剂对C2H4的生成显示出98.9%的选择性,生成速率为 67.65 μmol g –1 h –1。 该工作“Engineering the Charge Density on an In2.77S4/Porous Organic Polymer Hybrid Photocatalyst for CO2‑to-Ethylene Conversion Reaction“为题发表在《Journal of the American Chemical Society》上。
【研究亮点】
1. In2.77S4尖晶石的两种不同氧化态实现了C-C偶联过程;
2. In2.77S4和POP之间形成了Z机制,C2H4的形成速率与任何其他报告相比,在光催化领域是最高的(图 1)。
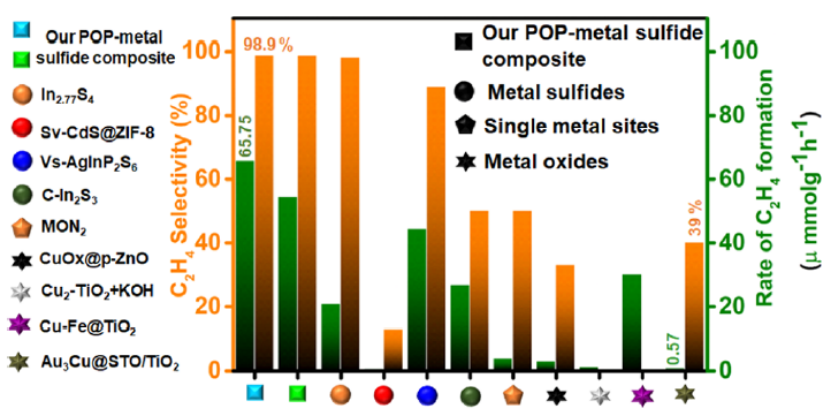
图 1.本研究与文献的性能比较。
【图文导读】
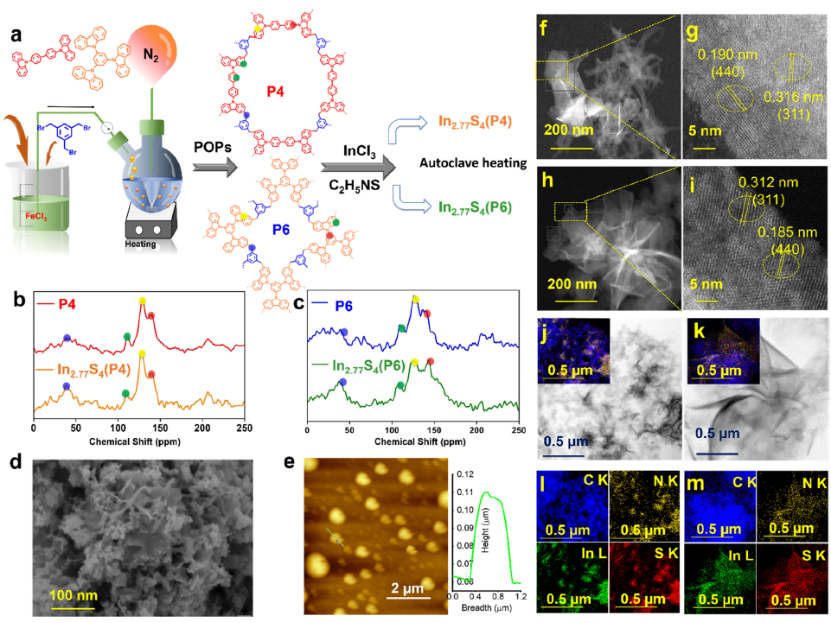
图 2.(a)采用Friedel-Crafts烷基化和溶剂热法合成基于咔唑的P4、P6、In2.77 S4 (P4)和In2.77S4 (P6) 。(b, c) 13 C CP/MAS-NMR谱图。(d)原始In2.77S4 (P6)的FESEM。(e) In2.77S4的AFM。In2.77S4(P4) (f, g) 和In 2.77 S4 (P6) (h, i)的 HAADF-STEM。In2.77S4(P4) (j, l) 和In2.77S4(P6) (k, m)的STEM-EDX。
合成POPs和In2.77S4 (P)多孔复合材料的合成示意图如图2 所示。P4和P6的13C CP MAS NMR(图 2 b、c)中位于~39 ppm 处的显着峰对应于 TBB的亚甲基碳原子,而位于~109.8 ppm 处的共振峰归因于聚合物骨架的未取代碳。在~125.7和139.8 ppm 处出现两个强共振峰归因于聚合物骨架中 BCB 和/或TCB单元的C取代和N取代碳。图 2 b、c的绿色曲线中的信号肩峰在 150–160 ppm 左右,这是由于多孔有机聚合物(P4和P6)的咔唑单元中存在“C=N”。 原子力显微镜 (AFM) 表明In2.77S4的高度约为 50 nm(图2e)。
In2.77S4 (P)的FE-SEM图像显示In2.77S4微花被微孔聚合物包裹(2d),在In2.77S4和PO之间形成异质结,电子转移通过该异质结进行。HAADF-STEM表明In2.77S4纳米花很好地分散在微孔聚合物基质上,表明聚合物的掺入不会改变In2.77S4异质结构(图2f-i)。In2.77S4(P)催化剂的 HAADF 元素映射(图 2j–m) 显示In和S主要位于被聚合物包裹的某些特定位置。
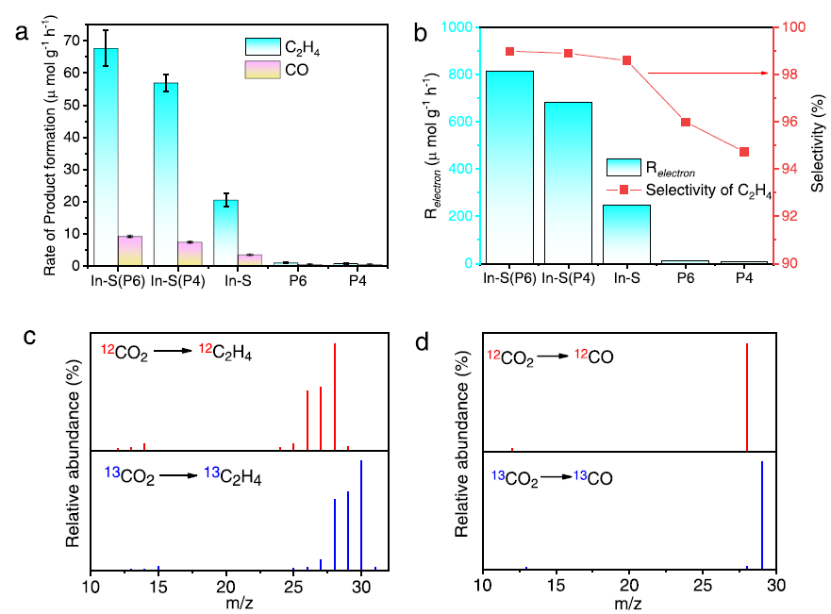
图 3. (a) 反应10小时的光催化CO2转化率。(b) 反应电子数和选择性。(c, d)同位素标记实验。
如图3a所示,C2H4生产率显示In2.77S4是CO2加氢的关键活性物质,表现出20.7 μmol g –1 h –1 C2H4生产率。另一方面,P6和P4没有显示出显着的 CO2还原性能。如图 3b所示,In2.77S4 (P6)具有98.9% 的C2H4选择性。同时,同位素实验表明C2H4(图3c)和 CO(图3d)来自CO2。
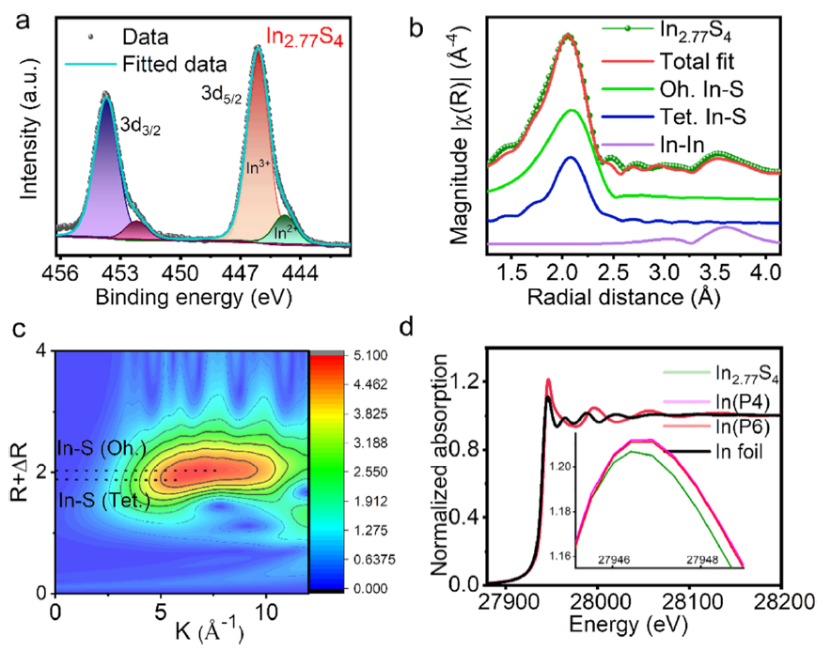
图 4. (a) In2.77S4的XPS。(b) In2.77S4的K边傅立叶变换EXAFS光谱。(c) 对In2.77S4的 k2加权EXAFS。(d) 在In2.77S4、In2.77S4(P4) 和In2.77S4(P6)的K边XANES光谱。
X 射线光电子能谱 (XPS) 表明存在 In2+和 In3+(图 4a)。图4b显示由于In-S散射而产生的明显第一层信号,而在较长距离处,强度较低的In-In第二层信号清晰可见。EXAFS数据的小波变换显示出一个加宽的点而不是一个集中的单点(图4c),表明存在两种不同类型的In-S键。
图4d中的In K边X射线吸收近边缘光谱 (XANES)显示了原始In2.77S4与具有P6和P4的复合材料之间的相似性。与原始In2.77S4相比,In2.77S4(P4)和In2.77S4(P6)中的白线强度有所增加表明复合材料中In中心的电子密度较低。这表明电荷从In2.77S4转移到有机物。
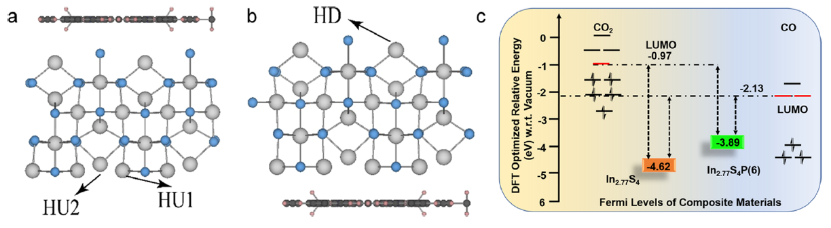
图 5. In2.77S4/POP异质结构的理论模型考虑了POP在In2.77S4的(a)上侧(异质向上 (HU))和 (b) 下侧(异质向下 (HD))吸附位置异质结构,其中灰色、天蓝色、黑色、粉红色和蓝色圆圈分别代表 In、S、C、H 和 N 原子。(c) CO2和CO LUMO位置相对于In2.77S4和In2.77S4(P6)的费米能级的示意图。
POP分别与In2.77S4的上表面和下表面相互作用(图 5a、b)。这些异质结构的Bader电荷计算表明,与 HD 相比,HU中的电荷分离更高,其中In2.77S4在每种情况下,表面都会向POP提供电子。In2.77S4和In2.77S4(P6)的费米能级分别位于-4.62和-3.89 eV(图5c)。CO和CO2的LUMO分别为-2.13和-0.97 eV,比复合材料更远离In2.77S4的费米能级。因此,从异质结构到吸附质的电子转移大于从原始In2.77S4的电子转移(图5c)。这是因为POP增加了In2.77S4/POP异质结构的总电子密度。
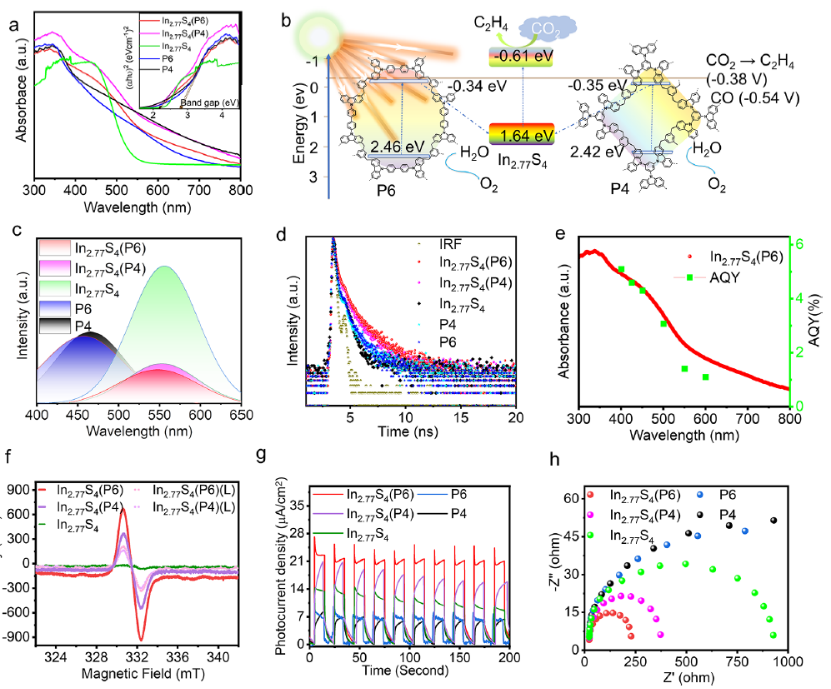
图 6. (a) UV-Vis DRS)。(b) In2.77S4/POPs的能带结构。(c)In2.77S4/POPs的稳态荧光光谱。(d) In2.77S4/POPs的TRPL。(e) AQY%。(f)ESR光谱。(g)光电流。(h) EIS。
图6中的UV-DRS光谱表明,本研究中的光催化剂具有出色的可见光捕获能力。与原始 POPs 相比,复合材料中的阶梯状光学吸收尾部可归因于In2.77S4吸收边。此外,在复合材料形成后,吸收峰高度增加,表明复合材料具有更好的光捕获能力。
P6、P4和In2.77S4的带隙分别为 2.8、2.77 和 2.25 eV。如图6b所示,两种POP的CBM均高于将CO2转化为C2H4或CO所需的最低电位。图6c显示了原始POP及其与In2.77S4的复合材料的稳态光致发光 (PL) 发射光谱。相对于原始 POP,In2.77S4/POP异质结构的PL强度显着降低。图6d显示复合催化剂的光生电荷平均寿命明显长于原始POPs或In2.77S4,这意味着复合材料中界面电荷转移更容易。
图6e表明在500 nm的激发下,AQY%符合吸收光谱。原位EPR实验中强信号主要是由于POPs中存在未成对电子,这些电子被In2.77S4的空穴湮灭,导致在光照后EPR信号强度降低(图6f)。复合材料具有更高的光电流密度和更小的电化学阻抗(图 6g,h)。
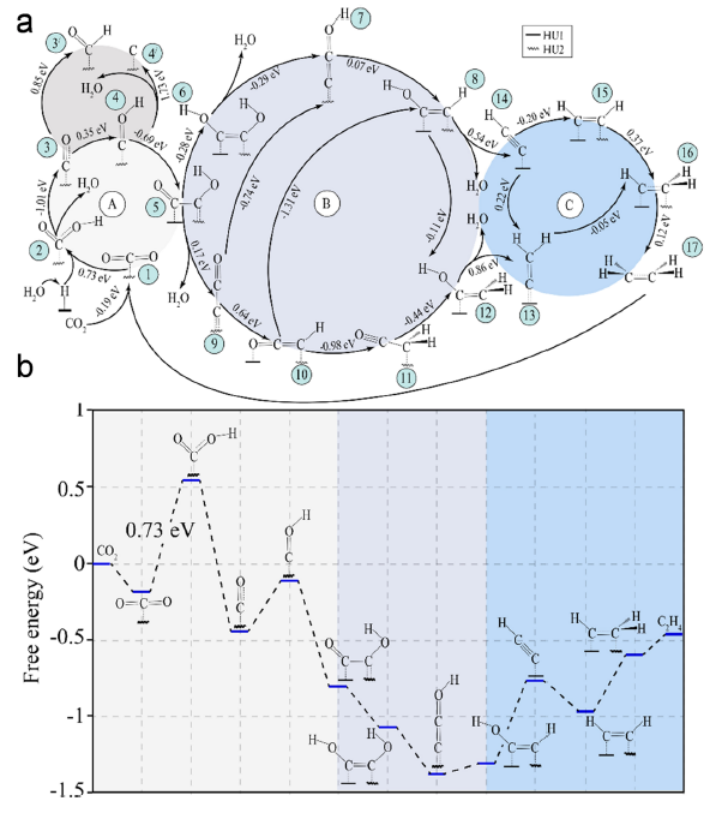
图 7. (a) C2H4生成的途径以及中间体之间相应的自由能变化。(b) In2.77S4(P)表面上形成C2H4的自由能图。
*COH与吸附在HU1上的 *CO 偶联形成 *OCCOH(图7a)是整个反应的决速步。由于*OHCCOH 与*C2O 相比具有稳定性,因此选择第二条途径(图7b)作为在电荷极化“In”中心形成C2H4的反应机制。
【总结与展望】
作者开发了一种光活性In2.77S4纳米花和POP基的多孔光复合材料In2.77S4(P) ,它在光照下选择性地将CO2转化为乙烯。XPS和XAS结果表明In2.77S4中存在不同电荷的In原子。In2.77S4(P)表面四配位的In2+和六配位的In3+在CO2到多碳 (C2+) 产物转化中起着至关重要的作用。此外,POPs和In2.77S4形成了Z型机制增强了整体CO2光还原性能。本研究提供了在可见光下完成CO2到多碳 (C2+) 产物转化的新范例。
审核编辑:刘清
-
反向电子转移!双-单原子催化剂助力CO2光还原2023-08-29 3219
-
CO2辅助生成富含晶界的Cu催化剂实现高效CO-CO偶联2023-03-17 2105
-
分子催化剂助力酸性条件下的CO2还原2023-01-10 3085
-
如何更好地报道CO2电还原的性能?2023-01-08 2858
-
在熔盐中利用液态金属锡阴极实现整体碳中和电化学还原CO22022-12-30 3336
-
揭示卤素掺杂Sn基催化剂促进CO2电还原制甲酸盐原因2022-12-29 4219
-
红磷负载Au单原子实现CO2光还原为C2H62022-12-05 2960
-
CO2转化为C2产物的高效光催化剂的设计研究2022-11-25 4643
-
通过Cl-吸附在Ag HF电极上使CO2电还原为CO的效率2022-10-18 2459
-
蜂窝状多孔结晶异质电催化剂实现高效的CO2吸附/活化2022-09-30 4274
-
利用结构依赖性电化学特性来引导CO2还原途径2022-09-06 1923
-
一种用于低浓度CO2的有效光还原的技术2022-09-02 2969
-
聚合物电池的分类,锂聚合物电池的结构2009-05-24 4180
全部0条评论

快来发表一下你的评论吧 !
