

DS-PAW scf自洽计算的内容
描述
第一性原理平面波密度泛函计算软件DS-PAW是Device Studio平台下的一款使用C++开发的国产第一性原理密度泛函计算软件,使用平面波作为基函数组,其赝势是使用投影缀加平面波方法构造的。
DS-PAW能够应用于不同场景,例如金属、半导体、绝缘体、表面、磁性、非磁性和锂电等;能够精确预测材料的电子分布;能够进行原子几何结构优化;能够广泛的应用于材料科学领域。
本期将给大家介绍DS-PAW scf自洽计算的内容。
自洽计算能够得到特定晶体的电荷密度和波函数,有了电荷密度之后才能有计算该体系的能带、态密度等电子结构性质。特别需要注意是:自洽与能带、态密度等电子结构性质计算是有先后顺序的,必须先进行自洽计算得到电荷密度才能进一步计算能带、态密度等电子结构性质。
2.2.1. �� 原子自洽计算之准备输入文件
输入文件包含参数文件 scf.in 和结构文件 structure.as , scf.in 如下:

scf.in 输入参数介绍:
可以看到 scf.in 的输入文件中很多参数与结构弛豫的参数名是一致的,其设置方法也是一致的,这里只着重介绍一些前面没设置过或设置有些不同的参数:
task :本次计算为 scf 自洽计算,因此将task设置为 scf ;
cal.cutoffFactor :表示截断能参数 cal.cutoff 的系数,在DS-PAW程序中,如果 cal.cutoff 参数缺失,程序将根据元素的截断能设置默认的平面波截断, cal.cutoffFactor 参数就是在 cal.cutoff 上设置乘以一个系数;
io.charge :当io.charge设置为 true 时,表示计算完成之后输出电荷密度的二进制文件rho.bin和json文件rho.json,二进制rho.bin文件用于后续的后处理计算,例如能带、态密度等,rho.json文件用于显示;
io.wave :当io.wave设置为 true 时,表示计算完成之后输出波函数的二进制文件wave.bin,用户可以在后续的后处理计算中选择是否使用wave.bin开始计算;
structure.as 文件参考如下:
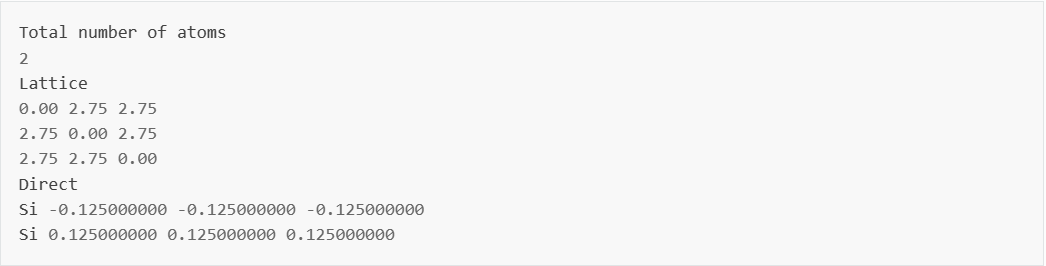
由于本案例为自洽计算只是为了计算得到特定体系的电子结构,因此不需要手动改变原子位置;
注解
io类参数只在结构弛豫和自洽中起作用,例如io.charge在其他计算情况下将不会生成rho.bin或rho.json文件;
在结构弛豫和自洽中,还能够保存elf、potential的数据,只需要将io.elf和io.potential设置为true即可;
如果用户想要使用自己优化的结构,只需在计算中将sys.structure参数指定绝对路径或相对路径下的relax.json,也可以将relax.json文件复制到本次计算的目录中,设置sys.structure=./relax.json即可;
带自旋体系的计算案例详解第二章的NiO案例。
计算时如需给体系添加背景电荷,可直接设置sys.electron参数,该参数指定价电子的总数。
2.2.2. run程序运行
准备好输入文件 scf.in 和 structure.as 后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in 。
2.2.3. analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 system.json 、 rho.bin 、 rho.json 、 wave.bin 这5个文件。
rho.bin :电荷密度的二进制文件,用于后续的后处理计算;
rho.json :电荷密度的 json 格式文件,用来显示电荷密度的结果;
wave.bin :波函数的二进制文件,用于后续计算;
使用 Device Studio 可直接对 rho.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 rho.json 即可,可根据作图要求自定义设置面板参数,处理可得一维、二维、三维电荷密度图,其中三维图如下所示:
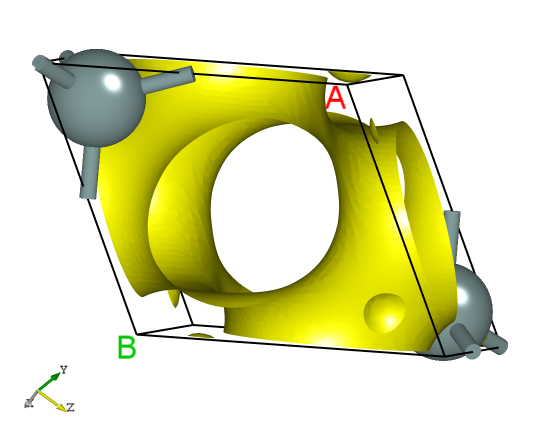
另可使用python脚本将 rho.json 格式的转化成 VESTA 软件支持的格式,具体操作见 辅助工具使用教程 部分。
。
-
 jf_17270223
2023-10-10
0 回复 举报请问 DS-PAW计算三维空间的能带结构 全空间撒点的话 band.kpointsLabel和band.kpointsCoord、band.kpointsNumber怎样设置呢 收起回复
jf_17270223
2023-10-10
0 回复 举报请问 DS-PAW计算三维空间的能带结构 全空间撒点的话 band.kpointsLabel和band.kpointsCoord、band.kpointsNumber怎样设置呢 收起回复
-
DS-PAW bader电荷计算2023-02-27 3877
-
DS-PAW bandunfolding能带反折叠计算2023-02-25 3223
-
介绍DS-PAW efield外加电场计算的内容2023-02-22 2924
-
介绍DS-PAW aimd分子动力学模拟的内容2023-02-21 4476
-
DS-PAW vdw范德瓦尔斯修正计算2023-02-11 3468
-
DS-PAW hse杂化泛函计算2023-02-10 4359
-
DS-PAW pcharge部分电荷密度计算2023-02-09 2690
-
DS-PAW potential势函数计算2023-02-06 2622
-
DS-PAW band能带计算2023-02-02 2774
-
DS-PAW应用实例2022-08-11 3775
-
Device Studio应用实例之DS-PAW2022-08-10 4168
全部0条评论

快来发表一下你的评论吧 !
