

串联催化加速一氧化碳的转移和溢流实现CO2电还原为乙烯
描述
【研究背景】
化石燃料的过度使用导致了大量的二氧化碳排放,引发了能源和环境危机。二氧化碳转化为其他增值化学品是循环经济的基石。二氧化碳电还原反应是促进全球碳资源利用的重要途径之一。CO2ER可以生产各种含碳产品,其中乙烯(C2H4)是CO2ER的重要产品,可用于制造塑料和纤维等商品材料。Cu因其适当的*CO偶联强度和对*H中间体的弱吸附能力,被广泛认为是CO2ER生成C2H4最有效的催化剂之一。CO2活化的高能垒和CO2转化为C2H4的较正起始电位阻碍了其在CO2ER电催化中的广泛应用。
因此,需要设计兼具较强CO2活化能力和*CO偶联能力的催化剂。一个很有前途的策略是设计由两种不同催化剂组成的串联体系,同时提高活性和选择性。虽然串联催化剂催化CO2ER已经取得了一定的进展,但由于电合成C2H4中*CO转移过程的动力学缓慢,严重阻碍了它们的实际应用。
因此,设计一种能大量供应*CO的串联催化剂,以增强*CO转移动力学,提高*CO在Cu催化剂表面的覆盖率,是可取的方案。另一方面,采用氢气溢流策略,通过加速二元组分催化剂中*H中间体的转移。由此产生了一种同时促进*CO转移和溢流的策略,通过在串联催化剂上设计串联的CO分子管理,产生的CO首先解吸到电解质中,然后在另一组分上重新吸附,这种提出的直接*CO转移和溢流策略(图1)此前从未用于CO2ER电催化。
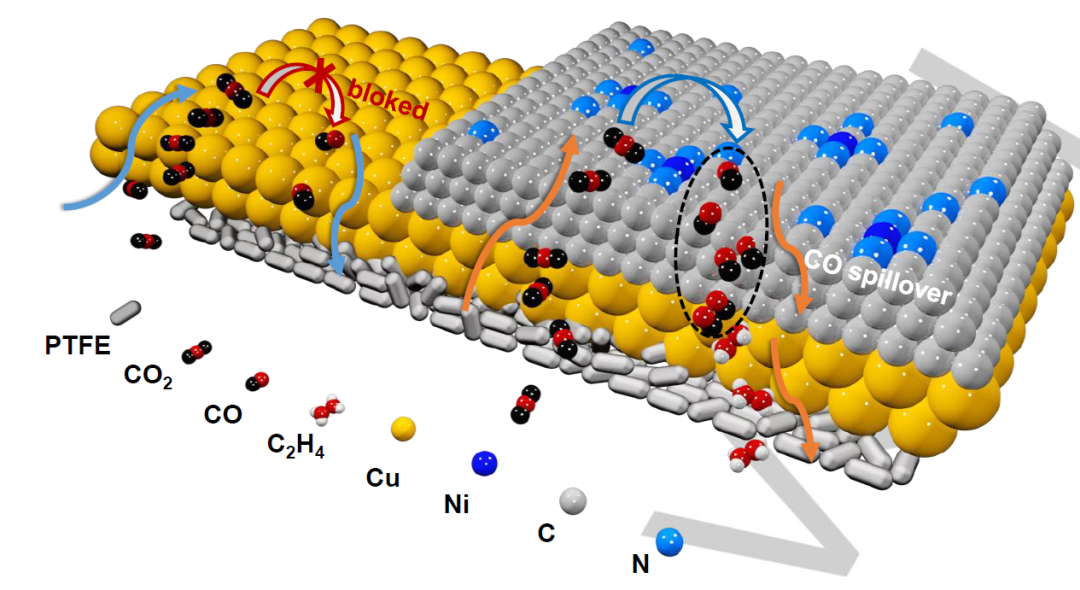
图 1.Cu NPs+Ni-SOD/NC的*CO溢流催化机理。
【成果简介】
浙江大学侯阳(通讯作者)开发了一种原位*CO生成和溢流策略,通过将单个Ni原子负载在具有辉锌矿(SOD)拓扑结构的富吡啶碳(Ni-SOD/NC)载体上,可以将*CO中间体提供给相邻的Cu纳米颗粒(NPs)。在-0.72 V的低电位下,C2H4选择性为62.5%,电流密度为160 mA cm-2。该工作以“Accelerated Transfer and Spillover of Carbon Monoxide through Tandem Catalysis for Kinetics-boosted Ethylene Electrosynthesis “为题发表在《Angewandte Chemie International Edition》上。
【研究亮点】
1. 具有吡啶N基团的NiN3活性位点加速*COOH中间体的形成,促进了*CO的解吸; 2. Cu NPs表面的溢流增加了*CO中间体在Cu NPs表面的覆盖度,提高了生成C2H4的选择性。
【图文导读】
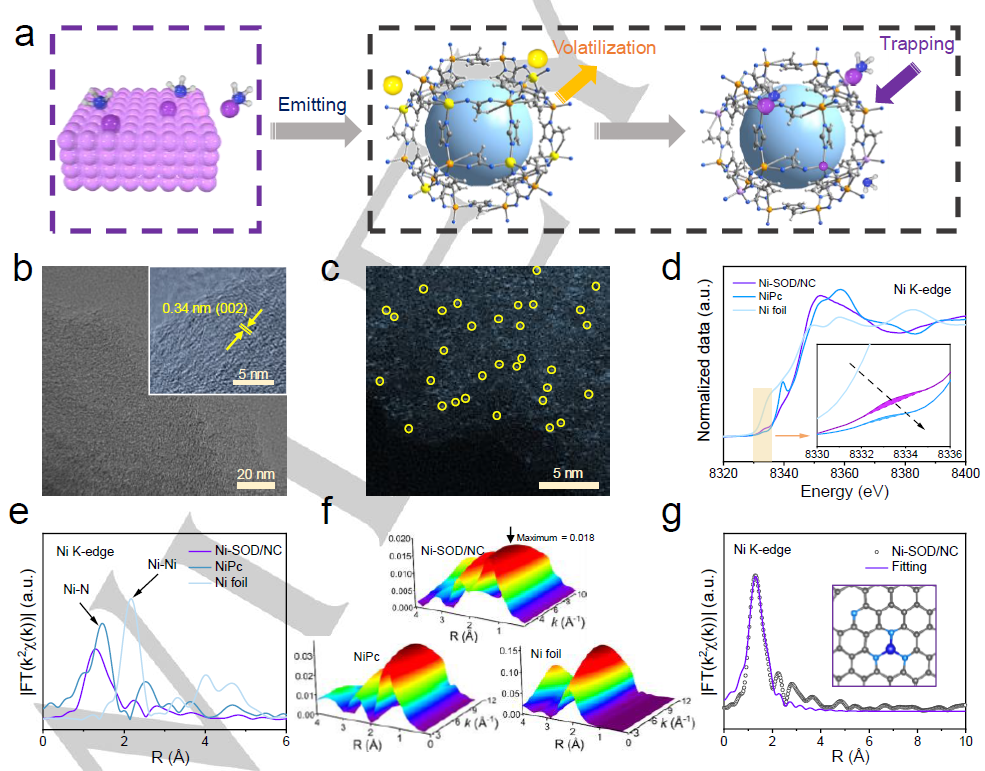
图 2. (a) Ni-SOD/NC合成示意图。(b) Ni-SOD/NC的TEM。(c) Ni-SOD/NC的 HAADF-STEM (d) Ni K边 XANES。(e) EXAFS谱的k2加权χ(k)函数。(f) WT-EXAFS光谱。(g)傅里叶变换EXAFS。
通过将大块Ni直接转化为Ni单原子,采用发射俘获策略制备Ni-SOD/NC催化剂,如图2a所示。TEM (图2)的图像显示其为十二面体,没有形成额外的金属NPs,高分辨率TEM (HRTEM)图像显示了晶格空间为0.34 nm的石墨条纹 (图2b)。HAADF-STEM显示Ni物种原子分散在碳载体上,大小为~ 0.25 nm(图2c)。XANES光谱显示Ni-SOD/NC的谱线强度介于Ni箔和镍酞菁(NiPc)之间(图2d)。Ni-SOD/NC在1.3 Å处表现出Ni-N配位 (图2e),与NiPc相比,其峰值强度较低,R距离较小,表明Ni-N配位环境不同。
此外,两种样品在2.2 Å处均未检测到Ni-Ni特征峰,证实了高度分散孤立的Ni原子的存在。小波变换(WT)(图2f)显示Ni-SOD/NC的WT最大值(0.018)位于4.8 Å-1,这归因于Ni-N键,但没有Ni-Ni峰。为了更清楚地识别N的贡献,得到了Ni-SOD/NC的R空间拟合曲线(图2g),与实验结果吻合较好;曲线拟合结果表明,Ni-N配位属于单原子Ni-N3位点(图2g)。与NiPc中Ni-N4的配位数相比,Ni-SOD/NC中Ni-N3的配位数较低可能是由于相对较高的热解温度和氨环境。
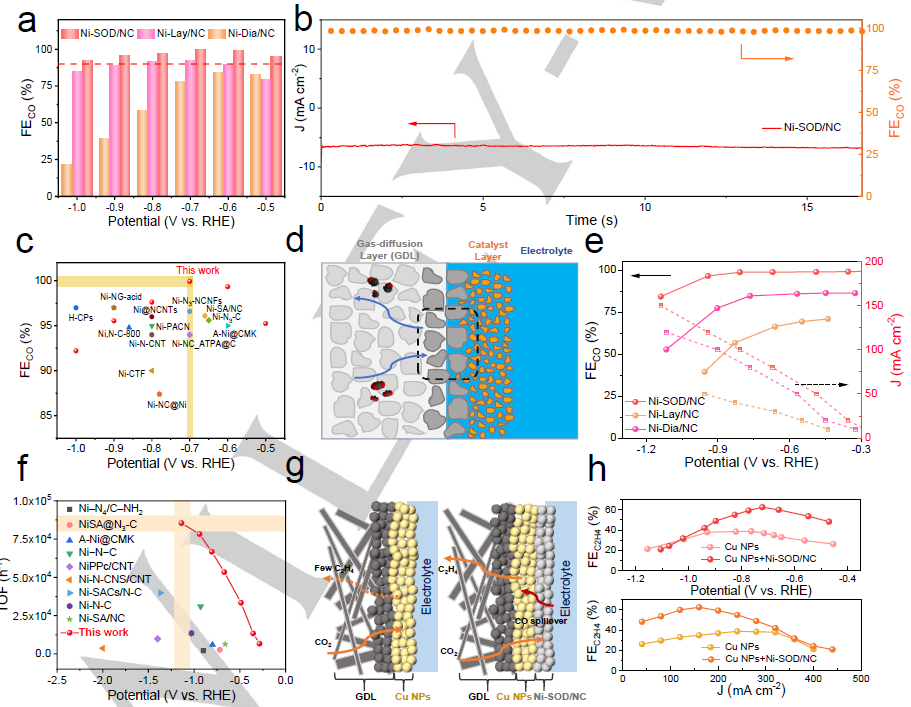
图 3. (a)Ni/NC的FECO。(b) Ni-SOD/NC在-0.7 V下的电流密度和FECO (c) H池中的FECO。(d)流动池示意图。(e) 流动池中Ni/NC在不同电位下的FECO和(j)总电流密度。(f)与其他报道的原子分散Ni/NC催化剂相比,Ni- SOD/NC在流动池中的TOF。(g) C2H4电合成Cu NPs和Cu NPs+Ni-SOD/NC工艺示意图。(h)不同电位和电流密度下Cu NPs和Cu NPs+Ni-SOD/NC的FEC2H4。
在-0.7 V时,FECO在99.9%以上(图3a)。Ni-SOD/NC催化剂在电催化16小时期间的电流密度和FECO的长期稳定性测试表明,Ni-SOD/NC催化剂具有较高的CO2ER稳定性(图3b)。与其他已报道的CO2ER催化剂(包括原子分散Ni/NC)相比(图3c),新开发的Ni-SOD/NC表现出出色的CO2-CO转换性能,在-0.7 V下FECO的最高选择性为99.9%,这保证了后续C2H4电合成良好的*CO生成和转移能力。为了克服溶解CO2在水溶液中的质量迁移限制,构建了CO2ER流动池(图3d),其中气体扩散层(GDL)作为Ni-SOD/NC催化材料的载体。
在流动池中,CO2通过气相快速扩散到催化剂,有利于高电流密度和快速传质。如图3e所示,Ni-SOD/NC在-0.94 V、120 mA cm-2(工业级电流密度)的FECO高达96.5%。Ni-SOD/NC的TOF在-0.94 V下最高可达85,398 h-1(图3f)。鉴于Ni-SOD/NC催化剂具有优异的CO2ER-CO性能,进一步采用CO管理策略,将自进料Ni-SOD/NC产生的*CO中间体外溢到Cu表面上,提高Cu催化剂表面的*CO覆盖率,进而促进CO2ER的*CO偶联。以新开发的Ni-SOD/NC催化剂为供体,在CuNPs上加料*CO,在聚四氟乙烯(PTFE)纳米纤维表面溅射沉积一层CuNPs,制备了CuNPs+Ni-SOD/NC催化剂(图3g)。
如图3h所示,当外加电位为-0.72 V,电流密度为160 mA cm-2时,Cu NPs+Ni-SOD/NC产生C2H4 FE最高,为62.5%。Cu NPs+Ni-SOD/NC的FEC2H4值(62.5%)是纯Cu NPs FEC2H4的1.62倍,在-0.45 V至-0.80 V的更宽电位范围内,前者的FEC2H4值比后者高出1.0倍以上(图3h)。相对于Ni-SOD/NC, CuNPs+Ni-SOD/NC上除了FEH2略有增加外,FECO急剧下降,而FEC2H4明显增加,这说明Ni-SOD/NC上生成的*CO溢出到CuNPs表面,进一步形成C2H4。
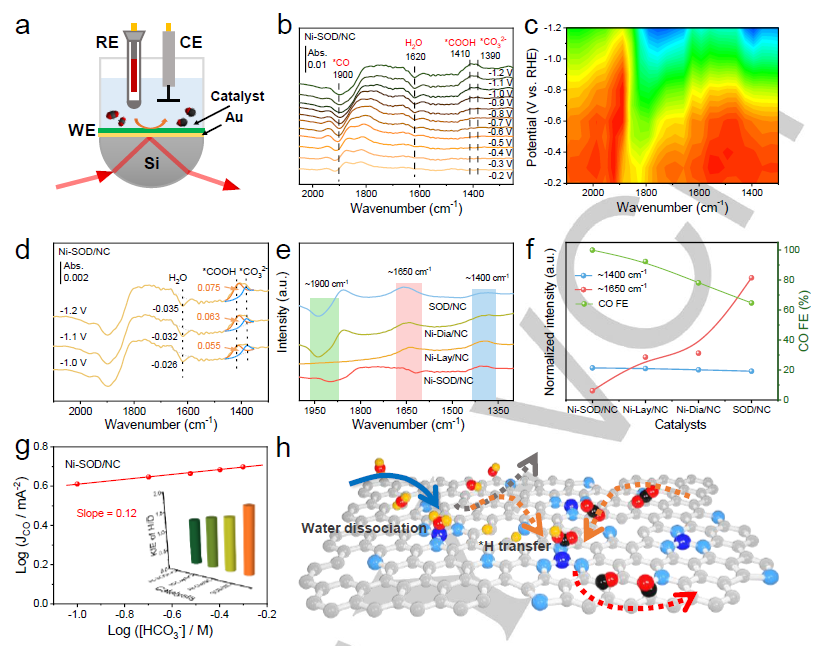
图 4.(a) ATR-FTIR原位测试装置。(b) Ni-SOD/NC的ATR-FTIR原位光谱。(c) Ni-SOD/NC ATR-FTIR原位光谱等高线图。(d) Ni-SOD/NC从-1.0 V至-1.2 V的ATR-FTIR原位光谱。(e) Ni-NC系列和SOD/NC在-1.2 V时的ATR-FTIR原位光谱。(f) Ni-NC系列和SOD/NC在-1.2 V时的峰面积及其对应的最大FECO。(g) -0.7 V下不同KHCO3浓度下Ni-SOD/NC的Jco。(h) Ni-SOD/NC对CO2ER的催化机理。
作者用原位ATR-FTIR测试确定CO2ER中间体(图4a)。图4b,c显示,位于1900、1620、1410和1390 cm-1的波段分别属于*CO、吸附的H2O、*COOH中间体和*CO32-中间体。在1620 cm-1处的峰来自吸附水,随电位的增加而负移,这是由于连续消耗H2O分子,并向新形成的*CO2提供质子,促进了快速质子化形成*COOH。在1410 cm-1附近出现了一个宽的正峰,归因于吸附的*COOH (图4d)。比较了SOD/NC、Ni-SOD/NC、Ni-Lay/NNC和Ni-Dia/NC在-1.2 V下的原位ATRFTIR (图4e)。图4f表明,H2O峰的积分面积与FECO成反比,而*COOH峰的积分面积与FECO成正比。
该结果进一步表明,水解离生成的*H加速了*COOH的生成,从而增强了CO2ER动力学,促进了CO的生成。Ni-SOD/NC在-0.7 V下的JCO随KHCO3浓度的增加呈现了0.12的近零斜率(图4g),说明形成*COOH中间体的质子不是来自KHCO3,而是来自水解离。 为了进一步验证Ni-SOD/NC在CO2ER过程中通过水活化和质子化过程加速了加氢动力学,作者在SOD/NC、Ni-SOD/NC、Ni-Lay/NC和Ni-Dia/NC上测量了H/D的CO生成速率和动力学同位素效应(KIE)(图4g),反映了电解质中水解离的质子转移速率。Ni-SOD/NC的KIE值为1.19,低于SOD/NC(1.76)、Ni-Lay/NC(1.32)和Ni-Dia/NC(1.41),说明水解离不是Ni-SOD/NC的速率决定步骤。Ni-SOD/NC中Ni-N3位点和吡啶N的存在通过促进水解离,加速了质子转移,如图4h所示。
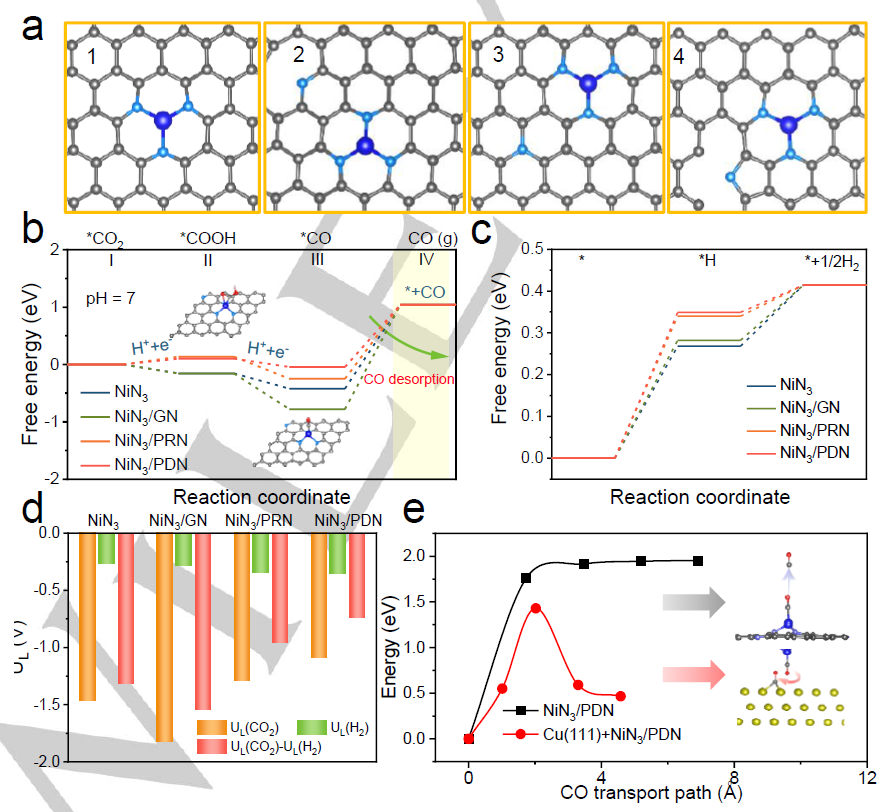
图 5. (a) NiN3、NiN3/PDN、NiN3/GN和NiN3/PRN的原子结构。(b)NiN3、NiN3/PDN、NiN3/GN和NiN3/PRN的CO2ER路径的自由能和结构演化。(c)HER过程中的自由能图。(d)CO2ER和HER之间的决速步电位差。(e)*CO解吸的能垒。
基于上述研究,作者构建了四种可能的模型,分别为NiN3、NiN3/GN、NiN3/PRN和NiN3/PDN (GN、PRN和PDN分别代表石墨氮、吡咯氮和吡啶氮)(图5a)。如图5b所示,计算得到NiN3/PRN和NiN3/PDN模型从*CO2转变为*COOH中间体的自由能分别为0.14和0.10 eV。对于NiN3、NiN3/GN、NiN3/PRN和NiN3/PDN模型,从*CO2转变为*COOH的反应能垒分别为2.12、1.92、1.91和1.74 eV,结合自由能结果,证明吡啶N比吡咯N更有利于*CO质子化形成*COOH,这与原位ATR-FTIR结果吻合。此外,由*COOH转化为*CO所计算的自由能均呈下降趋势。
从*COOH到*CO的反应势垒分别为0.68,0.70,1.32和0.01 eV,这也表明NiN3/PDN是所有构型中反应活性最高的产物。此外,NiN3、NiN3/GN、NiN3/PRN从*CO到CO的上坡自由能分别为1.46、1.81和1.29 eV,均远高于NiN3/PDN的上坡自由能(1.09 eV),说明附近的吡啶N也促进了*CO解吸步骤,有利于CO2ER。从图5c可以看出,与NiN3 (0.27 eV)、NiN3/GN (0.28eV)、NiN3/PRN (0.34 eV)相比,NiN3/PDN (0.35eV)的产氢受到明显抑制。此外,将UL定义为UL(CO2)-UL(H2)的差值,UL作为CO2ER选择性的描述符进行计算,根据这一定义,一个更正的值表示对二氧化碳还原的更高选择性。
在所有研究模型中,NiN3/PDN的UL(CO2)-UL(H2)最高(图5d),表明CO选择性最高。如图5e所示,*CO从NiN3/PDN解吸转移到Cu表面(图5e下半部分)的能垒明显低于从NiN3/PDN直接解吸进入气相(图5e上半部分),这进一步证明了Ni-SOD/NC解吸增加了*CO在Cu表面的覆盖面积,从而促进了C2H4的形成。因此,可以推断Ni-SOD/NC上产生了大量的*CO。
【总结与展望】
该工作通过设计一种由原子分散的NiN3锚定在吡啶富氮碳载体上的单Ni原子催化剂,验证了一种提高CO2ER活性的*CO转移和溢流策略。新开发的Ni-SOD/NC具有出色的*CO供应能力,在高达120 mA cm-2的工业级电流密度下,FECO约为96.5%,并且具有出色的稳定性。与吡啶N相邻的孤立的NiN3位点加速了*CO2到*COOH的质子化动力学,并降低了后续*CO解吸的能垒。*CO从Ni-SOD/NC层外溢流到CuNPs,在-0.45 V至-0.95 V的宽电位范围内,通过CO2ER选择性生成C2H4,导致FEC2H4达到62.5%。这项工作不仅为CO2ER和HER之间的*CO供给策略提供了深入的理解,而且为探索CO2ER及其他串联催化剂开辟了新的途径。
审核编辑:刘清
-
便携式一氧化碳检测控制仪2013-11-17 5
-
基于stm32一氧化碳检测系统2017-11-30 5782
-
一氧化碳传感器在汽车上的应用2019-07-22 2020
-
造成一氧化碳传感器故障的原因是什么?2021-06-16 2129
-
超低功耗一氧化碳检测仪的解决方案2022-09-21 1321
-
基于单片机的一氧化碳检测系统设计2023-09-25 904
-
一氧化碳检测仪的原理_一氧化碳检测仪的使用2017-12-04 15206
-
一氧化碳报警器的分类_一氧化碳报警器原理2019-08-06 5234
-
地下车库一氧化碳CO浓度监控系统安装说明2020-04-17 15168
-
一氧化碳探测器多久需要校正一次?2021-10-12 3345
-
一氧化碳检测仪能检测甲烷吗?-欧森杰2023-01-04 988
-
一氧化碳检测仪有哪几种采样方式?2023-01-09 1527
-
一氧化碳常见的几大误区2021-12-02 1533
-
产品选型 | 一氧化碳传感器选型指南2021-11-08 3000
-
地下车库一氧化碳浓度为多少时启动一氧化碳监测系统2023-08-24 1971
全部0条评论

快来发表一下你的评论吧 !
